
More Information
Submitted: June 12, 2024 | Approved: June 26, 2024 | Published: June 27, 2024
How to cite this article: Sforzin I, Beal JR, Moura F. Molecular Mechanisms and Potential Predictive Biomarkers in Advanced Non-small Cell Lung Cancer: A Summary of Current and Future Trends. Arch Surg Clin Res. 2024; 8: 039-061.
DOI: 10.29328/journal.ascr.1001082
Copyright License: © Sforzin I, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Non-small-cell lung cancer; Biomarkers; Signaling pathways; Targeted therapies; Precision medicine

Molecular Mechanisms and Potential Predictive Biomarkers in Advanced Non-small Cell Lung Cancer: A Summary of Current and Future Trends
Isabella Sforzin1*, Juliana Rodrigues Beal2 and Fernando Moura2
1Faculdade Israelita de Ciências da Saúde Albert Einstein – FICSAE, Avenida Padre Lebret, São Paulo - 05653-120, Brazil
2Hospital Israelita Albert Einstein, Avenida Albert Einstein 627/701 - São Paulo 05652-900, Brazil
*Address for Correspondence: Isabella Sforzin, Faculdade Israelita de Ciências da Saúde Albert Einstein – FICSAE, Avenida Padre Lebret, São Paulo - 05653-120, Brazil, Email: [email protected]
Non-small-cell lung cancer (NSCLC) accounts for 85% of lung cancer cases and is associated with different risk factors (smoking habits, gender, and age). In this scenario, many studies have been conducted to pursue improvement of survival, faster and better therapy response, reduced adverse events, and expanded available therapies and treatments against tumor resistance to drugs. These studies have focused on defining the most prevalent NSCLC biomarkers (EGFR, HER2, ALK, MET, ROS1, BRAF, KRAS G12C, HER3, NTRK, and NRG1) and their actionability. It is noteworthy that expressed kinase receptors can have overlapping mechanisms of activation of different pathways (JAK-STAT, MAPK, PI3K-AKT-mTOR, and PLC-c), which can lead to the same outcome of cell proliferation, migration, and survival resulting in increased tumor resistance to treatment. This review provides an overview of the latest findings regarding NSCLC treatment, emphasizing particular biomarkers and potential molecularly altered pathways implicated as targeted therapies. Additionally, it explores the clinical significance of the proposed treatments, their implication on progression-free survival, ongoing clinical trials, and their perspective of evolution so far.
NSCLC: Non-Small Cell Lung Cancer; SCLC: Small-Cell Lung Cancer; ORR: Objective Response Rate; PFS: Progression-Free Survival; EGFR: Epidermal Growth Factor Receptor; HER: Human Epidermal Growth Receptor; ALK: Anaplastic Lymphoma Kinase; MET: Mesenchymal Epithelial Transition; ROS1: Receptor Proto-Oncogene; BRAF: V-Raf Murine Sarcoma Viral Oncogene Homolog B1; MET: Rearrangement during Transfection; KRAS: Kirsten Rat Sarcoma 2 viral oncogene homolog; NTRK: Neurotrophic Tropomyosin-Related Kinase; NRG1: Neuregulin; JAK: Janus-Activated Kinase; STAT: Signal Transducer and Activator of Transcription; MAPK: Mitogen-Activated Protein Kinase; PI3K:Phosphatidylinositol 3-Kinase; AKT: Protein Kinase B; mTOR: Mammalian Target of Rapamycin; PLC: Phospholipase C; PKC: Protein Kinase C; TKI: Tyrosine Kinase Inhibitor; ORR: Overall Response Rate; Ex20ins: Exon 20 insertion; CPCT: Carboplatin-Pemetrexed Chemotherapy; DM1: cytotoxic agent Emtansine; TDM1: Trastuzumab-emtansine; DXd: Deruxtecan; TDX-d: Trastuzumab-Deruxtecan; CNS: Central Nervous System; EML4: Echinoderm Microtubule-associated protein-like 4; NPM1: Nucleophosmin 1; BCL11A: B-Cell Lymphoma/leukemia 11A; HGF: Hepatocyte Growth Factor; SF: Scatter Factor; CDK4: Cyclin-Dependent Kinase 4; MDM2: Murine double minute 2; IHC: Immunohistochemistry; FDA: Food and Drug Administration; RTK: Receptor Tyrosine Kinase; ADC: Antibody Drug Conjugates; CT: Chemotherapy; NF-kB: Factor Nuclear kappa B; MEK: Methyl Ethyl Ketone; TRK: Tropomyosin Receptor Kinase; GTP: Guanosine Triphosphate; ERKs: Extracellular Signal Regulated Kinases; IRS: Insulin Receptor Substrate; PTEN: Phosphatase and Tensin Homolog; RAS: RAT sarcoma; RAF: Rapidly Accelerated Fibrosarcoma; PDL1: Programmed cell Death Ligand 1; ELBD: Extracellular Ligand-Binding Domain (ELBD); TMD: Hydrophobic Transmembrane Domain; CTKD: Cytoplasmic Tyrosine Kinase Domain; EGF: Epidermal Growth Factor;TGFα: Transforming Growth Factor Alpha; CCD6: Coiled–Coil Domain-containing protein 6; NCOA4: Nuclear Receptor Coactivator 4; KIF5B: Kinesin Family Member 5b; TRIM33: Tripartite Motif-containing; NRTN: Neurturin; ARTN: Artemin; PSPN: Persephin; GDNG: Glial Cell line-derived neurotrophic factor; TGFb: Transforming Growth Factor beta
Amongst all types of cancer, lung cancer is one of the most frequent and major causes of mortality in developed countries. According to the World Health Organization (WHO), lung cancer can be further categorized as Small-cell lung cancer (SCLC), with a 15% prevalence rate, and Non-Small-Cell Lung Cancer (NSCLC), accounting for 85% of cases [1-3]. Risk factors predictive of increased incidence of lung cancer include age and behavioral components, such as the current tendency of electronic and non-electronic cigarette smoking, secondhand passive smoking, occupational exposure, and air pollution [1]. Within the three histological NSCLC subtypes (adenocarcinoma, squamous cell carcinoma, and large cell), NSCLC adenocarcinoma is the most prevalent, accounting for 40% of all patients with lung cancer [2]. Given the high prevalence of NSCLC, understanding and studying the molecular changes associated with tumor progression have enabled emerging options for targeted therapies towards a more personalized treatment approach, influencing the clinical course of the disease, and morbidity, and allowing for increased disease-free survival. Specific biomarkers have revolutionized and guided clinical treatment of this cancer spectrum, leading to the development of inhibitors that exhibit greater potency against their intended targets. Approximately 66% of NSCLC patients exhibit an oncogenic mutation, and about half of these patients have a lesion that can be therapeutically targeted, thus expanding treatment options [4-6].
Predictive biomarkers in NSCLC are essential for guiding personalized treatments and improving clinical outcomes for patients. They enable the identification of specific genetic mutations that respond to targeted therapies, thereby increasing the likelihood of successful outcomes. Additionally, they provide insights into resistance mechanisms associated with tumor progression and guide the selection of subsequent therapies. Biomarkers also offer prognostic information, helping to stratify patients based on expected disease progression and overall survival. These capabilities allow for a focus on therapies that are more likely to succeed, facilitating a more precise, effective, and personalized approach to treatment [1-6].
Although studies focused on understanding these biomarkers as potential targeted therapies are increasingly consolidated, treatment selection for NSCLC still faces significant challenges. The effectiveness of targeted therapies in NSCLC can be incomplete and transient due to factors of tumor heterogeneity, where various coexisting genetic mutations in cancer cells cause them to behave differently in response to each proposed treatment. When combined, these alterations can either generate sensitivity and response to therapy or, in the worst-case scenario, tumor resistance, whether intrinsic, adaptive, or acquired. Intrinsic resistance occurs in tumors that have mutations making them naturally naturally insensitive to available therapies. Adaptive response occurs when tumor cells undergo changes that allow them to survive and persist despite therapy. Acquired resistance results from a combination of different genetic factors within tumor cells combined with the development of new cellular changes during therapy [5].
The mechanisms by which tumors resist treatments can be classified as "on target," meaning when the initial target of a drug mutates, rendering it resistant and thus making the therapy ineffective, and "off-target" when alternative signaling pathways are activated, allowing the tumor to evade treatment and its cells to continue growing. The tumor microenvironment can also contribute as a resistance factor [5]. "Tumor tenacity" can be interpreted as the tumor's capacity to grow and spread even in adverse conditions (such as during drug action or immune system attacks), persisting and adapting within its environment. These mechanisms, primarily associated with gene activation, overexpression, amplification, and gene fusion or rearrangement, pose significant challenges to treatment. Finally, it is important to mention that the route of drug administration and differences in drug absorption, cytotoxic side effects, and the need for therapies that can cross the blood-brain barrier to penetrate and act on the central nervous system are additional challenges associated with therapeutic development [3-6].
In summary, NSCL is the most common subtype within the spectrum of malignant lung tumors and numerous studies are focused on validating specific treatments to improve survival. Within this context, the study of the main molecular mechanisms and their associated biomarkers shows promise for the development of targeted therapies that aim to contain tumor advancement. This review focuses on tumor pathways in NSCLC, including potential target biomarkers and their clinical relevance. Additionally, it discusses ongoing clinical trails and future perspectives.
Actionable biomarkers
In this review, the most prevalent specific biomarkers targeted by therapies were selected. Oncogenic mutations in the Epidermal Growth Factor Receptor (EGFR), Anaplastic Lymphoma Kinase (ALK), ROS1 proto-oncogene receptor tyrosine kinase (ROS1), and murine sarcoma viral oncogene homolog B1 (BRAF) are targets of medications that have been approved and consolidated by the US Food and Drug Administration (FDA) for the treatment of NSCLC.
Other oncogenic drivers, such as rearranged during transfection (RET), mesenchymal-epithelial transition (MET), Kirsten rat sarcoma viral oncogene homolog (KRAS), human epidermal growth factor receptor 2 (HER2), human epidermal growth factor receptor 3 (HER3), and neurotrophic tropomyosin receptor kinase (NTRK) were also selected for their impact as potential targets for targeted therapies. Each biomarker is presented due to its clinical relevance and potential impact on the treatment and prognosis of targeted therapies. Throught the text, we examine their molecular characteristics and therapeutic implications, considering mechanism of action, efficacy, response rate, prognosis, side effects, and tolerability.
The text below provides a comprehensive overview of drugs that could be used as targeted therapy for these biomarkers. They have either been assessed in predinical and/or clinical trials or are currently being studied. Drugs that have already received approval from the US FDA for use in NSCLC are listed in tables throught the text, along with their respective approvals for current use.
EGFR: EGFR, also known as HER1, is part of the HER family of receptor tyrosine kinases (RTKs), which includes EGFR (ErbB-1/HER1), HER2 (ErbB-2), HER3 (ErbB-3) and HER4 (ErbB-4) [7]. Oncogenic-driven EGFR-activation mutations are prevalent in advanced NSCLC adenocarcinoma histotypes, detected in approximately 20% of patients. Therefore, EGFR tyrosine kinase inhibitors (TKIs) are commonly used as a first-line treatment for the respective cancer, mainly focusing on specific alterations in the exons 18, 19, 20, and 21 of the EGFR gene that encodes the TK domain [5-8]. Monotherapy with EGFR TKIs showed good response and delayed occurrence of tumor resistance, ranging from 50% - 80%, resulting in improved progression-free survival [5]. These results are superior to those achieved with conventional chemotherapy [7]. The main inhibitors are divided according to the specific gene mutation and their correlation with EGFR signaling pathways.
First-generation EGFR-TKIs act by blocking the activation of the pathways through binding to the ATP docking site. Erlotinib and Gefitinib are representatives of this category. Due to the potential failure of the first-generation treatment, leading to tumoral acquired resistance, second-generation treatments, such as Afatinib and Dacomitinib, are also commonly used. Icotinib is another option still under research. It is important to mention that, regardless of the chosen therapy, the consequent development of resistance is inevitable. This is mainly related to developing resistance mutations in EGFR, HER2 amplification, or small-cell histological transformation. Therefore, investigations with third-generation selective EGFR TKIs for these mutations have been conducted. Among them, Lazertinib received its first approval in 2021 and continues to be evaluated for use in NSCLC, along with Olmutinib. Meanwhile, Osimertinib and Rociletinib have US FDA-approved uses [5-7,9].
Osimertinib is a third-generation TKI approved for the treatment of metastatic NSCLC with the T790M mutation progressing during or after therapy with other inhibitors. It is a medication that, in previous studies, administered as a second-line therapy, has shown superior CNS penetration compared to platinum chemotherapy. Preclinical and phase I data from the AURA trial are promising, with a median survival of 20.5 months for the use of the drug as first-line therapy for this EGFR mutation. The phase III FLAURA study evaluated the use of Osimertinib compared to Gefitinib or Erlotinib in treatment-naïve patients. A consistent benefit of Osimertinib in systemic and CNS response over standard EGFR-TKIs in terms of progression-free survival was demonstrated, with a similar safety profile, and lower rates of adverse events despite longer exposure to the drug [10]. In the case of disease progression involving other mutations, whether EGFR-dependent (such as C797S, L792X, G796X, and L718Q, which hinder the drug's binding to the ATP site), independent mutations (MET amplification), concomitant mutations, or those of unknown origin, the initial approach is usually made with Platinum-Based Chemotherapy (median PFS of 3.4 to 6.9 months) [11,12].
Due to the lack of targeted therapies or beneficial immunotherapies, additional treatments for disease control through alternative pathways are being studied. Among these,... is Amivantamab: a bispecific IgG antibody that binds to EGFR and MET. Based on phase I results of the CHRYSALIS study, it has received approval for the treatment of individuals with locally advanced or metastatic NSCLC featuring Exon 20 insertion (Ex20ins) in EGFR, whose condition progressed despite prior chemotherapy [12,13].
This trial is complemented by the ongoing CHRYSALIS 2 phase 1/1b study, which evaluates the response of EGFR mutations to the combination of Amivantamab and Lazertinib in two phases: monotherapy with dose escalation and dose expansion. In this second phase, different cohorts are further subdivided: A for EGFR ex19del and ex21 L858R mutations after Osimertinib and prior PBCT; B for EGFR exon20ins after platinum-based chemotherapy and standard therapy; C for atypical mutations after first or second-generation EGFR TKI therapy or treatment-naive; D aims to validate the strategy for EGFR ex19del or ex21 L858R patients naive to chemotherapy with recurrence after Osimertinib use [11-14].
Other studies were important to corroborate the assessment and effectiveness of this combined therapy. MARIPOSA-2 Phase III study focuses on evaluating the efficacy and safety of the combination of Amivantamab and carboplatin-pemetrexed chemotherapy or Amivantamab, Lazertinib, and chemotherapy versus chemotherapy alone in the treatment of Exon 19del or Exon 21 L858R substitution following the failure of monotherapy with Osimertinib. The hypothesis is that the positive synergistic effect for extracellular and catalytic domains of EGFR will lead to effective outcomes. As a result, both direct mutation or amplification and indirect EGFR/MET mechanisms of tumoral and polyclonal resistance were covered, with a good response rate and duration, including intracranial penetration [11-13,15]. PAPILLON phase 3 was conducted in patients with EGFR Ex20ins without prior systemic treatment, receiving intravenous Amivantamab plus carboplatin-pemetrexed versus chemotherapy [15].
CHRYSALIS pre-clinical assessments have shown potential prevention of ligand binding, internalization, and breakdown of receptors, as well as the activation of macrophages, monocytes, and natural killer cells via its Fc domain. The sum of these mechanisms represents a potential anticancer effect to circumvent MET and TK site resistance in mutated EGFR [11-13,15]. In all studies, side effects include skin rashes, paronychia, reversible cytopenia, and drug infusion-related reactions, all of which, in every case, had low clinical relevance. Venous thromboembolism is also noted, suggesting a potential need for anticoagulation. The PALOMA study (phases I and II) has been developing the subcutaneous formulation, pharmacokinetics, and safety dose of Amivantamab to reduce infusion-related adverse effects compared to subcutaneous administration. In the current state, this route of administration has shown better treatment tolerance [11,12,15,16].
The results have been promising. CHRYSALIS showed that the therapeutic combination´s efficacy against disease progression during or after Osimertinib suggests that the EGFR-MET blockade by Amivantamab can enhance the activity of Lazertinib as a first-line treatment [12]. Results from CHRYSALIS 2 corroborate these findings, including cohorts with relapse after Osimertinib and rescue chemotherapy therapy. MARIPOSA 2 Phase III suggests that both combined regimens are effective, with a similar average Progression-Free Survival (PFS), including intracranial PFS [11,12]. The same was observed in the PAPILLON study, which proved effective in the first-line treatment for Exon20del [15]. Finally, another potentially favorable association for similar benefits against mutant EGFR would be Osimertinib with Savolitinib or Tepotinib (targeting MET) [12]. Considering the premise of positive benefit suggested by CHRYSALIS, a study published in January 2023 was conducted to provide further external validation of Amivantamab's efficacy in real-world clinical practice. To achieve this, treatment databases primarily from Europe and the USA were evaluated (with TKIs being the mainstay). The outcome revealed robust evidence of statistically significant clinical benefit for Amivantamab in advanced NSCLC patients with EGFR Exon20ins mutations following platinum-based therapy [17].
It is important to note that resistance mechanisms to first-line treatment with Osimertinib are highly varied. In practice, identifying these mechanisms is often complex and uncertain. Therefore, another innovative treatment option worth mentioning would be HER3-targeted therapy, particularly considering that nearly two-thirds of NSCLC patient tumor cells express EGFR/HER3 positivity. Patritumab-deruxtecan (an antibody-drug conjugate) is being evaluated as a single agent for EGFR inhibitor-resistant NSCLC and will be further discussed [18].
HER2: Also known as ERBB2, HER2 is another member of the HER family of receptor tyrosine kinases and, as an oncogenic driver, observed in ~2% of NSCLC adenocarcinoma and related to patients with low or non-existent smoking history, female gender, oriental ethnicity and brain metastasis [6,19,20]. Recently, research has indicated that HER2 exon-20-insertion mutations, which result from the duplication or insertion of amino acids leading to protein overexpression, account for approximately 90% of NSCLC adenocarcinoma HER2 mutaions. This functional alteration potentially intensifies downstream signaling activation, leading to tumor development [6,20].
Many studies have demonstrated the effectiveness of targeted treatments for HER2. Over the years, the targeted therapies studied have focused on three groups: HER2 TKIs, anti-HER antibodies, or anti-HER2 antibody-drug conjugates [21,22]. The first treatments tested in NSCLC were those using dual inhibitors of HER2 and EGFR, such as Afatinib and Dacomitinib (pan-HER inhibitors), generally with insufficient and not consistently durable response. Neratinib (ErbB-receptor family blocker) for NSCLC is still under research [23-25]. Another example of a dual inhibitor, Lapatinib, still has limited evidence of effective action against mutated EGFR and HER2 [25].
Antibody therapy against HER2 protein includes Trastuzumab which targets various extracellular receptor domains and prevents extracellular molecules from activating the downstream pathway of dimerization of the ligands of the intracellular domain. Trastuzumab is a monoclonal antibody directed to the receptor and binds to the IV extracellular domain, resulting in HER2 shedding, tumor angiogenesis, and PI3K (Phosphoinositide-3 kinase) - AKT (Protein Kinase B) pathway inhibition, and cellular cytotoxicity. However, also due to the low prevalence of amplification or overexpression of HER2 proteins in NSCLC, clinical studies aimed at evaluating Trastuzumab in association with chemotherapy in this cancer spectrum have shown poor results of clinical benefit [20,23,26].
Yet, alternative therapies have shown promising results: when this antibody-drug is conjugated to the cytotoxic agent Emtansine (DM1) forming Trastuzumab-emtansine (TDM1), there is the possibility of its use for the treatment of HER2-positive cancers (approved for breast cancers) with potential resistance to conventional treatment with Trastuzumab. Thus, TDM1 can direct an acceptable cellular cytotoxic activity to tumor cells overexpressing the HER2 receptor, a change observed in about 20% of lung cancers [22-24]. Trastuzumab can also be conjugated to a topoisomerase I inhibitor forming the Trastuzumab-Deruxtecan (TDX-d) therapy, capable of delivering high levels of intracellular cytotoxicity in tumor cells, causing apoptosis induction and DNA degradation [20,22-24]. A recent study published in the New England Journal of Medicine showed the clinical potential of using this targeted therapeutic combination for HER-positive NSCLC. A 17.8-month overall survival after therapy was reported, indicating durable anticancer activity. However, it's important to note that the study did not evaluate cases of CNS metastasis and further research is needed to fully understand the effectiveness of this treatment approach [23].
Within therapeutic possibilities, HER-family TKIs can inhibit several TK receptors other than just HER2, such as HER 1 and HER4; PI3K inhibitors can directly inhibit the downstream signaling PI3K pathway; Fc-modified chimeric monoclonal antibody binds to HER2 causing the inhibition of tumor cell proliferation, reducing the shedding of the HER2 extracellular domain and mediating antibody-dependent cellular cytotoxicity [20,26,27]. Table 1 correlates each already US FDA-approved drug to its specific therapy.
| Table 1: Summary of EGFR and HER2 biomarkers associated with NSCLC resistance mutations and the respective drug (approved by US FDA) used to treat each of them. | |||
| Drug Name | Biomarker | Current FDA-approved indications (Source: fda.gov) | Reference |
| Gefitinib | Oral first-generation therapy EGFR TKI | First-line treatment for metastatic NSCLC with EGFR exon19 del. or exon 21 (L858R) substitution mutations. | [5-7,9] |
| Icotinib | Oral first-generation therapy selective EGFR TKI | Yet to be approved by the US FDA as a therapy for NSCLC. | [7] |
| Erlotinib | Oral first-generation therapy reversible EGFR TKI | (1) Second or third-line treatment; (2) Maintenance treatment of patients with locally advanced or metastatic NSCLC whose disease has not progressed after four cycles of platinum-based first-line chemotherapy; (3) Treatment of locally advanced or NSCLC after failure of at least one prior chemotherapy regimen. | [5-7,9] |
| Amivantamab | Bispecific antibody targeting EGFR and MET | (1) First-line treatment of locally advanced or metastatic NSCLC with EGFR exon20 insertion mutations; (2) Or whose disease has progressed on or after platinum-based chemotherapy. | [12-17] |
| Afatinib | Oral irreversible second-generation HER1, HER2 and HER4 inhibitor | First-line treatment of metastatic NSCLC tumors with non-resistant EGFR mutations. | [5-7,9,23-25] |
| Dacomitinib | Highly selective irreversible second-generation pan-HER inhibitor* | First-line treatment of metastatic NSCLC with EGFR exon19 del or exon 21 (L858R) substitution mutations. | [5-7,9,23-25] |
| Osimertinib | Third-generation prospective irreversible EGFR TKI | (1) First-line treatment and adjuvant therapy after tumor resection in patients with metastatic NSCLC whose tumors have EGFR exon19 del. or exon21 (L858R) mutations; (2) Treatment of metastatic EGFR T790M mutation-positive NSCLC whose disease has progressed on or after EGFR TKI therapy; (3) With platinum/based for patients with locally advanced or metastatic NSCLC whose tumor have EGFR exon19 del. or exon21 (L858R) mutations. | [5-7,14] |
| Lazertinib | Third-generation highly selective EGFR TKI with low toxicity and high CNS penetration | Yet to be approved by the US FDA as a therapy for NSCLC. | [5-7,9,11-15] |
| Rociletinib | Third-generation prospective reversible EGFR TKI | (1) Treatment of mutant EGFR in NSCLC that has been previously treated with an EGFR-targeted therapy; (2) and EGFR T790M mutation. | [5-7,9] |
| Olmutinib | Oral third-generation EGFR inhibitor | Yet to be approved by the US FDA as a therapy for NSCLC. | [5-7,9] |
| Dasatinib | SCR inhibitor in EGFR-resistant cell | Yet to be approved by the US FDA as a therapy for NSCLC. | [5-7,9] |
| Pertuzumab | Monoclonal antibody targeting HER2 | Yet to be approved by the US FDA as a therapy for NSCLC. | [22-24] |
| Trastuzumab | Monoclonal antibody targeting HER2 | Accelerated approval to fam-Trastuzumab Deruxtecan for treatment of patients with unresectable or metastatic HER2-positive solid tumors who have received prior systemic treatment and have no satisfactory alternative treatment options. | [20,22-24,26] |
| Trastuzumab - Deruxtecan | Anti-HER2 monoclonal antibody conjugated to topoisomerase I inhibitor | Accelerated approval to fam-Trastuzumab Deruxtecan for treatment of patients with unresectable or metastatic HER2-positive solid tumors who have received prior systemic treatment and have no satisfactory alternative treatment options. | [22-24] |
| Trastuzumab- Emtansine | Monoclonal antibody-drug conjugate targeting HER2 | Yet to be approved by the US FDA as a therapy for NSCLC. | [22-24] |
| Lapatinib | Oral dual reversible TKI | Yet to be approved by the US FDA as a therapy for NSCLC. | [25] |
| Neratinib | Oral irreversible HER-family TKI | Yet to be approved by the US FDA as a therapy for NSCLC. | [7,23-25] |
| *The hyperactivation of HER (erbB) family receptors, HER 1-4, represents significant targets for anticancer treatments. Therefore, studies focus on the development of small molecules capable of irreversibly blocking multiple HER receptors simultaneously: pan-HER inhibitors. T90M—secondary point mutation that substitutes methionine for threonine at amino acid position 790, C795S—secondary point mutation that substitutes cysteine for serine at amino acid position 795. **FDA-current approved therapies depend on the detection of the respective mutations by an FDA-approved test. | |||
ALK: The ALK gene is located on chromosome 2p23.2 [28]. It codes for a transmembrane enzymatic protein (ALK RTK or CD246) and can undergo three types of genomic alterations: rearrangement, amplification, and point mutation. ALK fusions result in an oncogenic receptor that can be identified in 5% of NSCLC [29]. This and other biomarkers' percentage of occurrence are specified in Graphic 1. Moreover, alterations in the ALK gene predominantly result from an inversion rearrangement with another gene that codes for fusion proteins, which pose a clinical concern due to their resitance to ALK TKIs. In NSCLC, especially in adenocarcinoma, the echinoderm microtubule-associated protein-like 4 (EML4) gene, can develop various overly expressed oncogene's fusions (EML4-ALK in 2% to 7% of tumors). Even though good sensitivity towards ALK TKIs between some fusion variants exists, recurring EML4-ALK protein is specifically a potential target for therapy in NSCLC treatment, such as for the use of Crizotinib [6,28-31].
Crizotinib is an ALK TKI, also known as an effective agent against ALK fusions. It is reported that its response rate has over 60% of success and that it can control 90% of installed tumors [28,31]. However, its use is also associated with tumor development and progression owing to drug resistance because of secondary ALK mutations [32]. Because of that, the concomitant use of other ALK TKIs (Ceritinib, Alectinib, Brigatinib, Lorlatinib, and Ensartinib) is indicated for Crizotinib-resistant patients, aiming for higher PFS rates. The first three drugs also have improved central nervous system (CNS) penetration [6,33]. Ensartinib's treatment - a new type of second-generation ALK inhibitor has been developed to enhance its effectiveness against metastases in the central nervous system - benefit results are seen both against resistant tumors and when more than two inhibitors are used [28].
Met oncogene: MET(The mesenchymal-epithelial transition) omcogene is located om chromosome 7q21-q31 and encodes MET receiver homologous to an RTK. This receptor is activated when binding to endogenous ligands, such as hepatocyte growth factor/scatter factor (HGF/SF) [34]. This process results in dimerization followed by phosphorylation of intracellular domains where other proteins bind, leading to activation of MAPK (Mitogen-activated protein kinase), PI3K=AKT and NF=kB (Nuclear factor kappa B) downstream signaling pathways. Due to this interaction between MET and these pathways, malignant tumor alterations in MET secondary to a tumor can impair regulatory components of intracellular signaling cascades. Examples of dysregulation include overexpression of MET or HGF protein, occurring in 35% - 72% of NSCLC cases due to abnormal MET mRNA levels; activation; MET gene amplification in 14% of NSCLC patients; deletion; mutation/rearrangement; and failure in MET gene transcriptional control. All these factors constitute an array of biomarkers that must be accurately assested for appropriate therapeutic decisions [6,35].
MET-target-therapies in NSCLC have shown better response in polytherapy with synergistic use of both MET and EGFR-TKIs [6,35,36]. As an example, this therapy is recommended for MET exon 14 loss mutations, mainly exon-14-skipping, that may occur concomitantly with CDK4 (Cyclin-dependent kinase 4), PI3K, MDM2 (Muring double minute 2) and EGFR mutations. METex14 mutation incidence varies in different histological types of NSCLC but approaches 3% - 4%, mainly, adenocarcinoma and adenosquamous carcinoma, and is mostly found in middle-aged non-smoker females.[35-40].
Comparative studies of combined therapies have linked the first-generation tyrosine kinase inhibitor Erlotinib with the non-selective tyrosine kinase inhibitor Tivantinib, concluding that the combination may be effective in patients with previously treated EGFR mutant and MET-positive NSCLC [41]. The multi-targeted kinase inhibitor Foretinib was recently evaluated in combination with erlotinib as an effective and safe combined therapy against advanced NSCLC that progressed after chemotherapy, achieving an Objective Response Rate (ORR) of 17.8%. Although this study suggests that MET status could serve as a biomarker for the use of this combination therapy in this study, these data still require further confirmation in the future [42].
Heterogeneity in the expression of MET within tumors implies a diversity of therapeutic agents to be used as targeted therapy. This includes monoclonal antibodies anti-MET (Onartuzumab, Emibetuzumab, and Telisutuzumab) and anti-HGF (Rilotumumab). Both antibodies are less utilized in cases of ligand-independent activation of MET and still require further elucidation of mechanism, efficacy, safety, tolerability, and prognosis. Also, there are the the TKIs, divided by on target type Ia crizontinib; type Ib Tepotinibn, Savolitinib, and Capmatinib and type II Merestinib, Glesatinib, and Cabozantinib. Of these, only Crizotinib, Tepotinib, and Capmatinib are effectively approved by the US FDA for NSCLC [38,39,42].
Crizotinib - US-FDA approved multikinase inbihitor for ALK and ROS1 - treatment's response rate was 50% for high-level MET amplification in NSCLC, being indicated as a first-line treatment superior to standard chemotherapy. Clinical response to this drug underscores the significance of studying protooncogenic mutations associated with MET as baseline biomarkers for developing inhibitors of the altered MET pathway. It is important to mention that second-side mutations can confer resitance to this drug. Moreover, despite being used against METex14 mutations, mainly off-label, studies have shown an average of 31% of ORR and around 9 months of median duration of response. Combining HGF/MET inhibition with targeted EGFR inhibitors represents a potential treatment for tumor resistance [35-40,43]. Table 2 correlates each already US FDA-approved drug to its specific therapy.
| Table 2: Summary of ALK, MET, and ROS1 biomarkers associated with NSCLC resistance mutations and the respective drug (approved by the US Food and Drug Administration) used to treat each of them. | |||
| Drug Name | Biomarker | Current FDA-approved indications (Source: fda.gov) | Reference |
| Ceritinib | ROS TKI and second-generation ALK TKI amid Crizotinib resistance | Frontline or Subsequent line treatment of patients with metastatic NSCLC whose tumors are ALK-positive or intolerant to Crizotinib. | [6,33,48,49] |
| Alectinib | Highly potent second-generation ALK TKI amid Crizotinib resistance | First-line or adjuvant treatment following tumor resection in patients with ALK-positive in NSCLC or Crizotinib resistant (better first-line option than Crizotinib). | [6,33] |
| Ensartinib | Potent ROS1 and ALK inhibitor (improved activity on CNS metastasis) | Frontline or Subsequent line treatment for metastatic ALK-positive NSCLC. | [28,33] |
| Brigatinib | ALK, EGFR, and ROS1 mutation inhibitor | Frontline or Subsequent line treatment for metastatic ALK-positive NSCLC and those who have progressed on or are intolerant to Crizotinib. | [6,33,48] |
| Lorlatinib | Highly potent third-generation ROS TKI and ALK TKI amid Crizotinib or next-generation TKI resistance | (1) Second or third-line treatment for metastatic ALK-positive NSCLC who progressed on Crizotinib and one another ALK TKI; (2) for disease progression after first-line treatment with Alectinib or Ceritinib. | [6,33,48,49] |
| Crizotinib | First ALK-targeted TKI, ROS1, and oral type Ia TKI for c/MET amplification | Frontline (Standard first-line oral TKI) or Subsequent line treatment for metastatic NSCLC ROS1-positive and ALK-positive tumors. | [6,28-31,35-36,38-40,42-44,46,48-49] |
| Capmatinib | Potential oral type Ib TKI for MET amplification, overexpression, and MET exon14 mutation and ATP-competitive inhibitor | Frontline or Subsequent line treatment for metastatic NSCLC with MET exon 14 skipping mutation. | [38,39,42] |
| Cabozantinib | Oral type II TKI for MET inactive DFGout conformation | Yet to be approved by the US FDA as a therapy for NSCLC. | [38,39,42] |
| Glesatinib | Oral type II TKI for MET inactive DFGout conformation | Yet to be approved by the US FDA as a therapy for NSCLC. | [38,39,42] |
| Meresetinib | Oral type II TKI for MET inactive DFGout conformation | Yet to be approved by the US FDA as a therapy for NSCLC. | [38,39] |
| Tepotinib | Selective oral type Ib TKI for MET amplification/overexpression and METex14 mutation | Second-line treatment for patients affected by NSCLC carrying MET exon 14 skipping mutations | [12,38,39,42] |
| Savolitinib | Selective oral Type Ib MET TKI under clinical development | Yet to be approved by the US FDA as a therapy for NSCLC. | [12,38-40] |
| Tivantinib | Non-selective TKI, Non-ATP-competitive MET kinase inhibitor for MET amplification and overexpression | Yet to be approved by the US FDA as a therapy for NSCLC. | [41] |
| Foretinib | Multi-target kinase inhibitor, angiogenesis, and cell proliferation blocker | Yet to be approved by the US FDA as a therapy for NSCLC. | [42] |
| Entrectinib | First-generation oral multikinase inhibitor tracking TRKA/B/C* - Also potent selective oral inhibitor against ROS mutations that cross the brain-blood barrier | (1) First-line treatment targeting ROS-1 and NTRK mutations in NSCLC; (2) preferred agent in associated brain metastasis. | [44] |
| Emibetuzumab | Anti-MET Monoclonal antibody that inhibits c-MET and HGF binding and inhibits the HGF/MET-dependent pathway overexpression | Yet to be approved by the US FDA as a therapy for NSCLC. | [39,42] |
| Telisotuzumab | Fresh approach as an antibody-drug conjugate targeting c-MET | Breakthrough therapy for patients with advanced or metastatic EGFR wild-type, nonsquamous NSCLC who (1) have high levels of c-Met overexpression and (2) whose disease has progressed on/after, platinum-based chemotherapy | [39,42] |
| Onartuzumab | Anti-MET monoclonal antibodies for MET amplification and overexpression, MET and HGF mRNA expression, circulating plasma HGF, and MET exon14 mutation. It disrupts the binding of the HGF alpha chain to the ligand binding domain of c-MET | Yet to be approved by the US FDA as a therapy for NSCLC. | [39,42] |
| Rilotumumab | Monoclonal antibody anti-HGF: inhibits c-MET and HGF binding and HGF/MET-dependent pathway overexpression | Yet to be approved by the US FDA as a therapy for NSCLC. | [42] |
| *The Neurotrophic tropomyosin receptor kinase (NTRK) comprises three transmembrane receptor tyrosine kinases, namely TRKA, TRKB, and TRKC. **FDA-current approved therapies depend on the detection of the respective mutations by an FDA-approved test. | |||
ROS1: ROS1 belongs to the subfamily of tyrosine kinase insulin receptors; ergo it has a substantial kinase domain and ATP binding site homology to ALK [44,45]. Also, both biomarkers are mostly found in young females who are light smokers [46]. Because of the structural similarity, it is granted that ALK inhibitors can be used as ROS1 target-therapy [6,45,46]. Although its physiological role is still not completely understood, it is known that around 1% - 2% of NSCLC cases can be affected by ROS1 gene fusions, with an estimated 10,000-15,000 new cases every year [44]. ROS1 fusion gene points are in exons 32, 34, and 35, containing the kinase domain. Because of that, ROS1 RTKs are TKI's targets [47]. Although Crizotinib therapy was first used as an ALK-TKI, due to its PFS estimated rates of 19.2 months, it was approved by the US-FDA as a cross-inhibitor and first-line therapy for ROS1 [6,44,46]. However, like tumor resistance development because of administered drug therapies, treatment failure due to ROS1 point mutations and signaling pathways bypassing disease progression may happen and overlap with those in ALK-rearranged [44]. The occurrence of brain metastasis in NSCLC ROS1-positive patients has been verified and requires the use of other TKIs that have better CNS penetration, such as Brigatinib (approved against ALK mutations), Ceritinib, Entrectinib, and Lorlatinib. These drugs are also evaluated as a therapeutic possibility in the presence of resistance to Crizotinib [48,49]. When there is also resistance through activation of bypass downstream pathways, ROS1 and other bypass RTK (such as Crizotinib plus EGFR inhibitor) therapies combined are needed. Lastly, other than immunotherapy and despite side effects resulting from chemotherapy (CT), both ROS sensitivity and response to pemetrexed CT can lead to almost 90% of disease control [44].
BRAF: The BRAF gene that encodes BRAF kinase protein can suffer mutation accounting for 1% - 4% of NSCLC. Those mutations occur mainly in exon 11 and exon 15 and can be divided into V600 (forming, mostly, monomers) and non-V600E (dimers) mutations. The first category includes BRAF-V600E (50% of prevalence) and the second one, BRAF-G469A/V (35%) and BRAF-D594G (6%). RAF (Rapidly accelerated fibrosarcoma) - type proteins are intermediates in the signaling cascade of the RAS and MAPK pathways and their mutations induce BRAF-monomeric downstream signaling. Although RAF inhibitors may reduce the affinity between dimers in non-V600 mutations, they do not inhibit them with the same efficacy as monomers, and worse yet, they can develop acquired resistance to dimers [6,29,50-53].
Vemurafenib and Dabrafenib are BRAF-V600-specific inhibitors used as a monotherapy or combined with both MEK (methyl ethyl ketone) and PI3K inhibitors, for outcome improvement [6]. Although BRAF and MEK inhibition is standard of care in this molecular subtype of NSCLC, it doesn't prevent the resistance development of bypass pathways. In addition, resistance to EGFR's therapy mechanisms has occurred because of BRAF mutations. The Dabrafenib plus Trametinib (MEK inhibitor) therapy was also FDA-approved against V600E and has induced a PFS of 10.2 months and an overall response rate of 67% [51,52,54].
KRAS: KRAS mutations are the most prevalent in Western countries, detected in 30% of NSCLC adenocarcinomas, and are rarely found in squamous cell cancers. Its counterparts NRAS and HRAS, however, have low mutation frequencies (≤ 1%) [56]. The majority of its mutations occur in codons 12 (80%) of chromosome 12p12.1 [57], the main ones being: the substitution of Glycine by Cysteine (KRAS-G12C) in 40% of cases; by Valine (KRAS-G12V), or by Aspartic acid (KRAS-G12D) [56,57]. In lung cancers, G12V is associated with a worse prognosis than G12D [58]. Less common are the mutations KRAS-G12A, KRAS-G12S, KRAS-G12R, and KRAS-G12F or in codons 13 (KRAS-G13C), 61 (KRAS-Q61H) [56,57], 63, 117, 119 and 146 [58]. Exposure to heavy smoking favors KRAS-G12C conversions [56,57]. Other co-mutations with KRAS include STK11 and KEAP1, both associated with a low response to platinum and docetaxel chemotherapy, as well as TP53, SMARCA4, CDKN2A/CDKN2B [56]. These mutations can coexist concurrently with EGFR and ALK mutations [57].
Due to its high affinity for GTP and the absence of a binding pocket on the surface of KRAS, it was long considered undruggable, and treatment remained focused on chemotherapy use [56,57]. However, recent discoveries regarding KRAS-G12C have led to its evolution into a predictive biomarker for response with targeted therapies. This mutation, in its inactive state, features both a small pocket (P2) and a side chain that allows binding with small molecule inhibitors. Thus, Sotorasib and Adagrasib have been developed (approved by the US-FDA for locally advanced or metastatic NSCLC with KRAS-G12C), along with Divarasib, JDQ443, and JNJ-74699157 (still in phases I and II studies) [56]. In the CodeBreak 100 study, Sotorasib showed an ORR of 37.1% and a PFS of 6.1 months. It was also approved for subsequent therapy for KRAS-G12C that persisted during or after treatment with platinum-based chemotherapy with or without immunotherapy [56,57]. In the CodeBreak 200 trial, it demonstrated a superior clinical response compared to docetaxel in previously treated patients [56]. The CodeBreak 201 trial is currently ongoing to evaluate it as a first-line therapy [56]. The Krystal-1 study demonstrated a positive response to Adagrasib monotherapy in previously treated KRAS-G12C patients with an ORR of 42.9% and a PFS of 6.5 months. Krystal-7 and Krystal-12 are ongoing to validate Adagrasib in the first line and in comparison with docetaxel, respectively [56,57]. Studies to evaluate these medications for co-mutated KRAS-G12C are currently underway [57].
Resistance to therapy with covalent KRAS-G12C inhibitors can occur through the activation of other genomic alterations such as BRAF, RET, ALK, and MET. Therefore, combination therapies targeting downstream and upstream KRAS pathways are used as indirect inhibition therapy [56,59]. An example is the pan-KRAS inhibitor BI 1701963, which blocks the Son of Sevenless-1 (SOS1) binding with KRAS-GDP [56,57], and inhibitors of Src homology 2-containing protein tyrosine phosphatase 2 (SHP2) (TNO155, GDC-1971) [56]. Alternative potential therapies include immune checkpoint inhibitors [56,57] (such as Pembrolizumab, which, despite showing clinical benefits in monotherapy, still requires further elucidation regarding its action with KRAS mutations) [57]; Vaccination therapy [56,57] (including mDC3/8-KRAS peptide vaccine, Long peptide vaccine targeting KRAS E, and mRNA-5671) [57], and T-cell response enhancement therapy [56,57].
The C-terminal portion of KRAS4A and KRAS4B variants containing farnesyl cysteine has also been proposed as a target for therapies against mutated KRAS. Salirasib is a farnesyl cysteine mimetic used to disrupt KRAS-membrane associations by competing with and preventing membrane binding of GTP-activated RAS. However, second-generation Salirasib, as well as Tipifarnib and Lonafarnib, which are farnesyltransferase inhibitors clinically evaluated, have shown limited clinical efficacy against KRAS prenylation [57,58,60].
KRAS is a common mutation with crosstalk signaling pathways through both PI3K and MEK. Therefore, the investigation of drugs targeting synergistic or downstream pathways of KRAS, such as PI3K-AKT-mTOR, and their co-directed inhibition is based on the development of therapeutic alternatives [56,57,61,62]. Initially, a study published in 2016 presented Pictilisib, a first-generation Pan Class I PI3K inhibitor, as a targeted therapy with a response against associated PI3K mutations (mutated PIK3CA and loss of PTEN). However, the development of resistance proved to be rapid. Apitolisib (second generation) showed a dual, non-statistically significant inhibition of mTOR and PI3K (in vitro). The dose-dependent antiproliferative effect for PIK3CA mutations was positive for both drugs [62].
Continued studies introduced Cobimetinib, a MEK inhibitor, which, in H1975 and A549 adenocarcinoma cell lines, once again induced lower cell proliferation. The combined treatment of these three drugs, although with variations among cell lines, generally exhibited a synergistic antiproliferative response and low cell viability, but insufficient cytotoxic effects. Nevertheless, it is important to understand that other concomitant mutations in simultaneous pathways may play a role in oncogenic changes and need further elucidation [62].
Within the line of treatment targeting MEK, there is Trametinib. However, for the treatment of patients with KRAS-mutated NSCLC, this drug showed a response rate similar to the conventional use of docetaxel CT [61]. Something similar was observed with Selumetinib treatment: despite its antitumor activity [57,59,60], its monotherapy did not show to be superior to docetaxel or pemetrexed-based CT [61]. Sorafenib (oral multikinase inhibitor directed at RAF), apart from good treatment results for recurrent or non-scaly refractory NSCLC, clinically does not demonstrate efficacy and is associated with adverse events [61].
HER3 receptor family: The assessment of the importance of the HER receptor family also extends to ERBB-3/HER3, a ligand-induced dimerization-activated tyrosine kinase receptor. The upregulation of HER3 expression has been observed as a factor of resistance to therapy against mutated EGFR and, therefore, a factor of poor prognosis. Whereas HER3 is expressed in 67% of NSCLC tumors, 10 to 15% of US patients, and 30 to 40% of Asian patients; HER3 mutations are positive in 2% of NSCLC (1% squamous and 1% adenocarcinoma) [18,63,64]. Multiple studies have been focusing on exploring HER-3 and associated mechanisms as targeted therapies that may potentialize EGFR-focused treatments [18,65].
Cancer treatments with antibody-drug conjugates (ADC) have been of great relevance in studies of patients with mutated HER3 and xenografts. Among those cases, mention is made of Patritumab Deruxtecan (HER3-Dxd) - a specific immunoglobulin G1 mAb antibody-drug formed by the conjugation of anti-HER3 antibody and topoisomerase I via tetrapeptide-based cleavable linker - used isolated or following Osimertinib pretreatment. This therapy has shown efficacy in muted-EGFR and EGFR inhibitor-resistant tumors (39% RR) [18,64]. Upregulated tumor lysosomal enzymes degrade endocytosed ADc, releasing cytotoxicity and cell death [64]. Although this mechanism has not yet been effectively approved, its functioning is similar to the already mentioned TDX-d and response in xenograft study models making this therapy potential for mutated and resistant NSCLC-EGFR.
Likewise, Zenocutuzumab (Zeno), is a specific antibody (bio-specific humanized immunoglobulin G1) associated with cellular cytotoxicity targeting HER2xHER3 cell surface extracellular domains. It is effective as high antibody concentration causes antibody-dependent cellular toxicity. Its dock-block mechanism consists of docking HER2 and blocking NRG1 to interact with the HER3 targeting arm, preventing its conformation that is necessary to the HER2-HER3 dimerization - thus, phosphorylation of HER3 intracellular domain and so on blocking downstream pathways (AKT, STAT3) [65]. Therefore, histological studies have shown sensitive Zeno therapy against HER2/HER3 locally advanced, metastatic, and unresectable tumors and its phase I and II studies are fast-track granted by the FDA [65] (Table 3).
| Table 3: Summary of BRAF and KRAS biomarkers associated with NSCLC resistance mutations and the respective drug (approved by the US Food and Drug Administration) used to treat each of them. | |||
| Drug Name | Biomarker | Current FDA-approved indications (Source: fda.gov) | Reference |
| Vemurafenib | BRAF-V600 mutation inhibitor | Yet to be approved by the US FDA as a therapy for NSCLC. | [50,51] |
| Dabrafenib | BRAF-V600 mutation inhibitor | Accelerated approval in combination with Trametinib for the treatment of unresectable or metastatic solid tumors (including NSCLC) with BRAF V600E mutation who have progressed following prior treatment and have no satisfactory alternative treatment options. *Not indicated for patients with wild/type BRAF solid tumors |
[6,51,52,54,59] |
| Trametinib | MEK/BRAF-V600 mutation inhibitor | Accelerated approval in combination with Dabrafenib for the treatment of unresectable or metastatic solid tumors (including NSCLC) with BRAF V600E mutation who have progressed following prior treatment and have no satisfactory alternative treatment options. | [51,52,54,61] |
| Selumetinib | MEK inhibitor | Yet to be approved by the US FDA as a therapy for NSCLC. | [59-61] |
| Sotorasib | KRAS-G12C inhibitor | Second-line treatment of NSCLC with KRAS G12C mutation in patients who have received at least one prior systemic therapy. | [56,57,59] |
| Adagrasib | KRAS-G12C inhibitor | Second-line treatment of NSCLC with KRAS G12C mutation in patients who have received at least one prior systemic therapy. | [56,57,59] |
| Pembrolizumab | KRAS indirect inhibition therapy - Humanized monoclonal antibody targeting PD1 | Adjuvant treatment following resection and platinum-based chemotherapy for stage IB, II, or IIIA NSCLC. | [57] |
| Salirasib | GTP-KRAS membrane binding inhibitor | Yet to be approved by the US FDA as a therapy for NSCLC. | [60] |
| Sorafenib | Oral multi-kinase inhibitor directed at RAF (treatment for recurrent or non-scaly refractory mutant KRAS) | Yet to be approved by the US FDA as a therapy for NSCLC. | [61] |
| Cobimetinib | Potent MEK inhibitor | Yet to be approved by the US FDA as a therapy for NSCLC. | [62] |
| Pictilisib | First Generation Pan Class I PI3K inhibitor | Yet to be approved by the US FDA as a therapy for NSCLC. | [62] |
| Apitolisib | Second Generation dual mTOR - PI3K inhibitor | Yet to be approved by the US FDA as a therapy for NSCLC. | [62] |
| Patritumab-Deruxtecan | ADC therapy; pre-therapy: Osimertinib | Yet to be approved by the US FDA as a therapy for NSCLC. | [64] |
| Zenocotuzumab | Common light chain bispecific Biclonics antibody targeting NRG1-HER3 | Yet to be approved by the US FDA as a therapy for NSCLC. | [65] |
NTRK: Advances in studies aimed at developing targeted therapies for NSCLC have progressed towards different genes, including the NTRK fusions, found in approximately 1% of patients. The NRTK gene includes NTRK1, NTRK2, and NTRK3, each encoding a specific tropomyosin receptor kinase, TRKA, TRKB, and TRKC respectively. NTRK has sevsral fusion parteners such as ETV6, LMNA, MPRIP and TPM3, SQSTM1 [5,66-68]. Knowing, therefore, that cancer cells depend on the super functioning of tyrosine kinases linked to the NTRK gene and that NTRK-rearrange cancers are sensitive to TRK inhibitors, this receptor has become a therapeutic target.
US FDA-approved Entrectinib (already mentioned, also as a ROS1 and ALK inhibitor) and Larotrectinib are first-generation tropomyosin receptor kinase (TRK) inhibitors in NTRK mutation-positive patients (Table 4) [5]. The antitumor activity of Entrectinib has been observed to stem from its ability to inhibit phosphorylation of fused TRK proteins, compete with ATP binding and so disrupt downstream signal conduction in mutated or metastatic NTRK solid tumors (LMNA-NTRK1 or ETV6-NTRK), within 57% ORR. In 2020, a study published in The Lancet reported that adverse events associated with the administration of 600 mg/daily are treatable or low-risk - such as overweight, fatigue, constipation, and anemia [69-72].
| Table 4: Summary of NTRK (1, 2, and 3), MET, and biomarkers associated with NSCLC resistance mutations and the respective drug (approved by US Food and Drug Administration) used to treat each of them. | |||
| Drug Name | Biomarker | Current FDA-approved indications (Source: fda.gov) | Reference |
| Larotrectinib | First-generation pan-TRKA/B/C selective inhibitor | Treatment of (1) solid metastatic tumors with NTRK-gene fusion without a known acquired resistance mutation;(2) or where surgical resection is likely to result in severe morbidity and have no satisfactory alternative treatments or that have progressed after treatment. | [5,68,71,73,74] |
| Taletrectinib | Second-generation highly selective ROS1/NTRK inhibitor | Yet to be approved by the US FDA as a therapy for NSCLC. | [5,75] |
| Repotrectinib | Second generation TRKA/B/C, ROS1, ALK and wild-type TRKA/B/C, ROS1 and ALK inhibitor | (1) First-line treatment of advanced or metastatic NSCLC with ROS1-positive mutation; (2) Second-line treatment for patients who previously received a ROS1-targeted drug (Crizotinib). | [5,72] |
| Selitrectinib | Second-generation TRKA/B/C inhibitor | Yet to be approved by the US FDA as a therapy for NSCLC. | [5,71,73,74] |
| Cabozantinib | Oral multikinase non selective inhibitor against RET; also tracks MET, ROS1, AXL, KIT, TIE2* | Yet to be approved by the US FDA as a therapy for NSCLC. | [81] |
| Vandetanib | Oral multikinase non-selectively targeting RET inhibitor; also tracks VEGFR and EGFR | Yet to be approved by the US FDA as a therapy for NSCLC. | [81] |
| Laventinib | Multikinase non-selectively targeting RET; also tracks KIT, VEGFR 1–3, PDGFRα, and FGFR 1–4** | Yet to be approved by the US FDA as a therapy for NSCLC. | [5,68-74] |
| Selpercatinib | Oral nano TKI inhibitors selectively target the RET kinase domain; including fused, point mutated, and resistant RET | Treatment for (1) locally advanced or metastatic solid tumors with RET gene fusion that have progressed on or following prior systemic treatment or (2) who have no satisfactory alternative treatment options. | [5,68,71,73,74] |
| Praseltinib | Highly potent selective RET inhibitor | Treatment for NSCLC with RET-positive gene fusions. | [5,75] |
| *AXL belongs to the TAM family of RTKs that regulates various physiological functions, such as cell proliferation, viability, adhesion, and movement. The name TAM stands for the first letter of its three constituents—Tyro3, Axl, and Mer. Tyrosine-protein kinase KIT is an RTK expressed on the surface of hematopoietic stem cells. Tie RTK plays a regulatory role in vascular homeostasis. ** FGFRs play a role in various biological processes, and their inhibition could potentially offer benefits against squamous NSCLC. However, the specific abnormalities of FGFRs in this type of cancer have not been extensively studied. VEGFR—Vascular Endothelial Growth receptor, PDGFRα —Platelet-derived growth factor receptor A, FGFR—Fibroblast Growth Factor receptors. | |||
Larotrectinib is a pan-highly selective orally bioavailable inhibitor for the three mentioned TRKs, whose mechanism of action includes blocking the binding of an ATP molecule to TRK, and so inhibiting the growth of cells that are positive for different NTRK 1/2/3 rearrangements and downstream RAF-MEK-ERK and PI3K-AKT pathways. Unlike Entrectinib, this drug has a lower CNS penetrance, but equally high ORR among solid tumors (75% rate) and potential control of adverse events - neutropenia, fatigue, cough, anemia, and increased alanine aminotransferase [68,71,73,74].
Despite NTRK-rearranged cancer's high sensitivity to TKIs targeted therapies with clinical efficacy, resistance invariably develops, especially after exposure to potent TKIs. This resistance can occur through the acquisition of mutations involving aminoacid substitutions and, as an example, solvent mutations are mentioned. Therefore, next-generation TRK inhibitors have been studied to combat these mutations and have demonstrated both in vitro and in vivo responses. These include Selitrectinib, Taletrectinib, and Repotrectinib [5][71-75]. Selitrectinib has activity mainly against TRK A/B/C domains secondary resistance mutations [5]; Repotrectinib was designed to inhibit TRK A/B/C, also ROS1 and ALK secondary kinase mutations [72] and Taletrectinib has a highly selective enzymatic and growth inhibition against ROS1 and NTRK1/2/3 mutations [75].
In summary, the NTRK on-target mutations discovered both before the start of the systemic therapy instituted and after demonstrated a good response to the use of Entrectinib and Larotrectinib alone or concomitantly with the therapy previously in use; Off-target mutations have shown a good response to the use of next-generation inhibitors in association with medications already prescribed for possible NSCLC mutations.
RET: The RET gene, located on the long arm of chromosome 10, encodes a transmembrane receptor tyrosine kinase. It is known that RET fusions with gained function are detected in around 1% - 2% of NSCLC tumors in females, young age (< 60), and non or light smokers [76,77]. In RET-positive lung cancer, the potential for developing brain metastasis can affect up to nearly half of patients, implying poor overall survival. Moreover, although intracranial responses are expected with the use of RET-targeted inhibitors, clinically, therapeutic outcomes remain poor, particularly when compared to the use of other inhibitors in ALK and ROS1-rearranged lung cancers. Several clinical trials are underway to investigate alternative therapies for metastatic RET with high CNS involvement. For instance, trial NCT01582191 in phase I aims to evaluate the combination of Vandetanib and Everolimus [78]. Among the activating RET fusions, coiled-coil domain-containing protein 6 (CCDC6)-RET, nuclear receptor coactivator 4 (NCOA4)-RET, kinesin family member 5b (KIF5B)-RET, and tripartite motif-containing 33 (TRIM33)-RET are mentioned [79,80]. RET rearrangements occur concurrently with the PI3K signaling pathway, MAPK effectors, and other TK (EGFR, ALK, HER2), and de novo fusions are associated with mutant-acquired resistance against anti-EGFR [81]. All these mechanisms will be better explained below.
There are several inhibitors used against RET mutations to date and are divided into Non-selective RET inhibitors (Cabozantinib, Vandetanib, and Lenvatinib) and Selective RET inhibitors (Selpercatinib and Pralsetinib) [81]. Other inhibitors are Sunitinib, Sorafenib, Alectinib, Divotinib, Potantinib, and Apatinib. Most of the evidence on the effectiveness of these drugs in lung cancer with RET rearrangement is derived from case reports and preclinical studies, including ongoing phases I and II. Selpercatinib is a RET kinase selective inhibitor with nanomolar potency against diverse alterations and CNS penetration [82]. Pralsetinib is a recently studied tolerable selective inhibitor for the treatment of patients with RET-positive fusion in NSCLC. The Arrow study - currently in phase I/II - demonstrated a 61% ORR for patients previously treated with platinum-based chemotherapy and 70% for treatment-naïve patients, even inducing intracranial activity, thus confirming Pralsetinib as first-line. Both confirmatory phase III studies, AcceleRET Lung (NCT04222972) and LIBRETTO-431 (NCT04194944), evaluating Pralsetinib and Selpercatinib versus standard therapy in advanced and metastatic NSCLC, respectively, are ongoing [83].
Phosphoinositide-3-kinase–protein kinase B (PI3K/PKB) and Mitogen-activated protein kinase (MAPK) pathways review
Receptors associated with enzymes are transmembrane proteins that pass through a plasmatic membrane connecting an extracellular domain to an intracellular one. Once connected to an extracellular molecule, this receptor can activate and transduce a specific cue that leads to the activation of an enzymatic activity connected to its internal domain. Those signal transduction-dependent pathways may regulate cell growth, proliferation, differentiation, and survival as a response to extracellular sign-protein called survival factors; cytoskeleton formation, and movement through non-diffusible signaling proteins, and phosphorylation by surface cell RTK cytoplasmic domain. The binding of a signal molecule in the form of as a dimer to an RTK extracellular domain provokes the association of two receptors forming a dimer, which propitiates the contact between its kinase domains in its intracellular tails, allowing multiple lateral protein phosphorylation. Each phosphorylated phosphatase tyrosine is a site to an intracellular molecule binding that propagates signals throughout the internal pathway, such as the Ras-GEF (guanine exchange factor) and PI3 kinase [4,84].
The signaling pathway PI3K-AKT-mTOR is correlated with tumor formation and progression. Its activation results from various mutations, such as in the PIK3CA gene (residues Glu 542, Glu 545, and HIS1047). These genetic alterations are identified in approximately 2 to 7% (in absolute numbers, 32 to 112 thousand patients) of lung cancer cases annually, and, in general, the overactivation of the PI3K pathway is associated with resistance to treatments such as chemotherapy, immunotherapy, and targeted therapies. This underscores the urgent need to develop effective therapeutic strategies and interventions that can benefit these patients. [4,62].
The MAP-kinase module activated by RAS: Ras-GEF binds to RTK athwart Grb2 (adapter protein) and switches Ras-GDP to Ras-GTP - activated transmembrane protein RAS form - that initiates de MAP (mitogen-activated protein) kinase signaling module. Ras must be inactivated so that de-cellular proliferation is controlled [4,84,85].
Initially, activated RAS recruits and activates MAPK kinase kinase (MAPKKK - including Raf, Mos, and Tpl2, each modulating a path) - via GTPase and phosphorylation proteins - that consequently phosphorylates MAPK kinase (MAPKK) which successive binds and activates in the same way MAPK. MAPK can be allocated into three groups: ERKs (extracellular signal-regulated kinases - ERK 1/2 and ERK 5); JNK/SAPK (Jun amino-terminal kinases/ stress-activated protein kinases) and p38 kinase (p38α, p38β, p38γ, and p38δ). Each subgroup drives an intracellular signaling cascade, with the Raf-MEK-ERK pathway being one of the most well-characterized MAPK signaling pathways. Those combined activated paths are responsible for cell growth, survival, differentiation, development, apoptosis, and inflammation control [4,84-86].
The multistep activation process of the PI3K pathway: As previously mentioned, PI3K binds to the transmembrane phosphorylated dimer protein via adapter molecules such as IRS (Insulin receptor substrate) and turns into its activated form, which is responsible for the conversion of its catalytic domain phosphatidylinositol bisphosphate (PI(3,4)P₂) - a cytoplasmic membrane-associated inositol phospholipid - to phosphatidylinositol trisphosphate (PI(3,4,5)P₃). The opposite conversion by dephosphorylation of PI3P is regulated by Phosphatase and Tensin Homolog deleted on Chromosome 10 (PTEN), which is therefore a tumor suppressor phosphatase active in the PI3K pathway. Signaling proteins such as PDL1 (Programmed cell death ligand 1) and PKB (protein kinase B, also known as AKT) that recognize this phospholipid are induced to bind to the membrane. Multiple kinase proteins recruited - PDK1 and mTOR - are responsible for AKT phosphorylation and, therefore, activation. Among its multiple functions, active AKT phosphorylates Bad (Bcl-2 associated agonist cell death) protein, turning it into its inactivated conformation and extricating activated Bcl-2, an apoptosis inhibitor [4,84,87]. The activation of mTOR and AKT and their extensive communication with other intracellular signaling pathways, as well as the loss of PTEN, are associated with an unfavorable prognosis. Regarding the PIK3CA gene, controversies persist about its role as an oncogenic driver or modulator of oncogenic effects, as well as its prognostic status [62].
Molecular mechanisms associated with mutations and tumoral resistance
EGFR: EGFR has three domains: the Extracellular Ligand-Binding Domain (ELBD), the hydrophobic transmembrane domain (TMD), and the Cytoplasmic Tyrosine Kinase Domain (CTKD). The ELBD binds extracellularly to Epidermal Growth Factor (EGF) and Transforming Growth Factor Alpha (TGFα), generating EGFR homo/ heterodimerization. Subsequently, it can activate other EGFR family RTK proteins that are bound to CTKD. This stimulates the intrinsic tyrosine kinase activity of the receptors that trigger the autophosphorylation of tyrosine residues. This signaling leads to the subsequent activation of downstream ATP-dependent pathways, including PI3K/ AKT) (also activated by SCR), rat sarcoma (RAS)/rapidly accelerated fibrosarcoma (RAF)/ MAPK) Janus kinase (JAK)/signal transducer and activator of transcription (STAT) and the phospholipase C-protein kinase C (PLC-PKC) pathway, that mediates various cellular processes, including cellular proliferation, differentiation, survival, and growth [6,84,88].
The main mechanisms associated with acquired tumor resistance to EGFR TKIs and responsible for the severity of NSCLC prognosis are i) oncogenic mutations that cause activation of various EGFR tyrosine kinase, which induces downstream signaling activation regardless of extracellular binding (constitutively active); ii) divergent activation of intracellular traditional EGFR signaling pathways via cognate receptors (hepatocyte growth factor receptor - MET, HER2), regardless of extracellularly binding of TFNα or EGF, enhancing cell proliferation, survival and inhibition of apoptosis; iii) histological transformations and mutations associated with the combination of first and second-generation therapies, amplifying the mentioned cellular processes [6]. Figure 1 shows signaling pathways that can be bypassed to achieve final processes that promote tumor resistance.
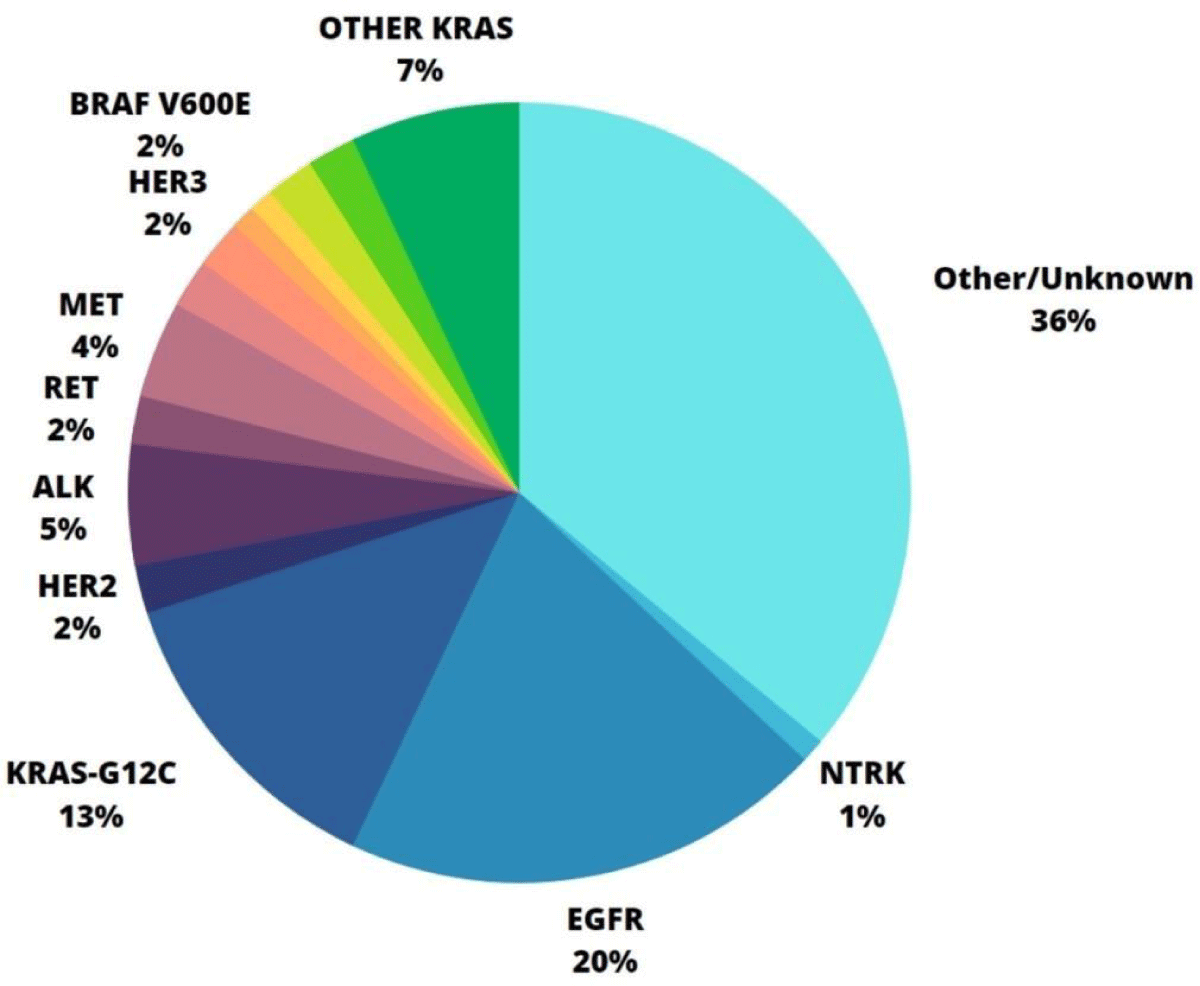
Figure 1: Phosphoinositide-3-kinase–protein kinase B (PI3K/PKB) and Mitogen-activated protein kinase (MAPK) pathways review. Percentage of occurrence of each biomarker in NSCLC adenocarcinoma [1-3,6-9,19,20,28-31,35,44,50-53,55-57,59,63,64-69,76,77].
HER2: HER2 is a membrane receptor coded by the homonymous gene located on chromosome 17 (17q12) and its oncogenic influence may occur in the form of gene amplification, protein overexpression, or chromosome 17 polysome. Structurally, the extracellular portion (ECD) of the receptor is composed of a ligand-binding domain that binds to extracellular molecules and promotes intracellular dimerization of HER2 molecules (homodimerization) or dimerization with another HER-family receptor (heterodimerization), which can bind to its tyrosine kinase domain (NH-CH-C terminal domain). This process leads to activation of downstream signaling of PI3K/AKT and RAS/MAP/MEK pathways. When heterodimerization occurs, HER2 and heterodimers exert a potent oncogenic signal [6,20,22,89]. This protein also mediates the sensitivity of EGFR-mutant lung tumors to anti-EGFR therapy [6]. HER3 and HER4 receptor activity occurs because of ligand binding to their extracellular region. Therefore, in the form of monomers, their dimerization and consequent activation of their dependent pathways are avoided. However, the HER2 inactive form does not exist: its dimerization occurs despite ligand binding [89].
The mechanisms of HER2-mediated tumorigenesis are an outcome of schematic signaling caused by HER2 overexpression that mainly leads to the formation of HER2-EGFR and HER2-HER3 dimers [6,20]. The increase in HER2-EGFR heterodimers provokes the intensification of downstream signaling duration (chiefly, MAPK pathway) due to the deviation of the EGFR endocytic degradation. Therefore, the recycling process of EGFR is diminished, which allows for its reutilization, re-binding, and activation as opposed to the intermittent interruption of its function that normally occurs [88]. HER2-HER3 heterodimers are primarily correlated to the oncogenic signaling of the PI3-AKT pathway because HER3 can bind more easily to the PI3K p85 subunit. Furthermore, PI3 activation depends on the phosphorylation of HER3 by HER2. Thus, HER2 overexpression can phosphorylate HER3 that is bound to PI3K resulting in its activation [90]. The latter (HER3/PI3K) can positively induce the tumor’s Akt functions, mainly apoptosis inhibition, cell growth, proliferation and response to nutrient availability, and angiogenesis [84,90].
ALK downstream pathways: The overactivation of the interconnected ALK downstream pathways (RAS-MAPK, PI3K-AKT, PLC-c) can be triggered by mutations in the ALK gene [30]. In addition, regardless of whether the overactivation is induced by rearrangement, fusion, overexpression of amplifications, or point mutations of ALK, it leads to cell proliferation, survival, and migration, conferring tumor persistence, mainly against ALK-TKIs in NSCLC [91,92]. This resistance can also result from two other mechanisms: i) ALK protein-independent or ii) ALK protein-dependent mechanisms. The first category includes both activation of bypass and downstream pathways through other biomarkers that have already been or will be discussed (EGFR, HER2, and MET) herein. The second category includes ALK kinase domain amplification, which may be correlated or not to normal gene alterations [6]. It is relevant to mention that ROS1 proto-oncogene RTK and ALK kinase domains’ resistance to TKIs happens because of their structural homology [30].
Superexpression and ligand-independent activation of ALK are partially determined by a translocation of a gene partner, forming a fusion gene that encodes oncogenic ALK proteins. However, despite the diversity of the partners (EML4, TGF, HIP1, TPR, PTPN3, KLC1, and KIF5B), they all retain the ALK kinase domain that leads to increased cell survival and proliferation as a result of the downstream signaling pathway activation [6,91]. The most common mutation product of tumor survival dependence, the EML4-ALK protein, results from the paracentric fusion of EML4 and ALK genes because of its opposite orientation in chromosome 2 [91]. The fusion points of EML4 are more variable than AL: they can occur in exon 13 (v1), exon 20 (v2), and exon 6 (v3). Regardless of which those points are, the EML-4 protein's N-terminal region fuses with the TK domain of the ALK gene and induces constitutive ALK kinase activity [92,93]. This activity is correlated to SRC (phosphorylates P13K), MAPK, and JAK-STAT pathways, by driving the phosphorylation of Akt, STAT3, and ERK. In summary, the importance of EML4-ALK v1 or v3 in the cell tumor being upstream to ERK lies in the fact that the apoptosis suppression in iL-3-dependent cell lines is overcome by ERK pre-phosphorylation by the EML4-ALK protein. Otherwise, the inhibition of ERK resulting from the expression of Bcl-2-like protein 11 (BIM) can induce cell apoptosis [84,93].
MET: Multiple MET gene dysregulation in tumors as mentioned before can be the result of different molecular mechanisms. In short, mutations related to the signaling pathway can be MET dependent (on target - overexpression or point mutations) or independent (bypass / off target), related to EGFR (as well as constitutive activation of downstream survival signals), HER, MAPK and KRAS mutations causing gene amplification, as described down below.
MET is a disulfide-linked homodimer made of large α- and small β-subunits; α- and the β- amino-terminal regions that form the extracellular domain (Sema domain) and the rest of the β-subunit forms a transmembrane and a cytoplasmic TK domain. MET can bind to HGF/SF through both NK1 and SPH domains. Activation depends on the phosphorylation of the catalytic or juxta-membrane (JM) domain via phospho-MET/Tyr1349. Due to that, missense and insertion mutations may happen in the semaphorin or JM domains, providing metastatic potential and aggressive phenotype in NSCLC adenocarcinoma [35,39,95-98]. Generally, cancer cells engage the tumor-associated fibroblast (TAF) to secrete in a paracrine manner the inactive precursor pro-HGF. The latter has its peptide that needs to be cleaved by the membrane-anchored enzyme matriptase on the cancer cell surface transforming it into active HGF [97]. Oncogenic mutations may exist in the Sema and juxtamembrane (R988C and T1010I) domains affecting receptor binding and increasing metastatic tumorigenicity respectively [39]. Nonetheless, this mechanism is not only related to NSCLC.
When HGF binds to its receptor, it triggers the homodimerization of two MET molecules and transphosphorylation on several binding sites on the catalytic domain, establishing both receptor activation and phosphorylation of other tyrosine residues (Tyr1349 and Tyr1356) inboard the docking site. The residues recruit and become docking sites for growth factor receptor-bound protein 2 (GRB2) and GRB2-associated binding protein 1 (GAB1); phospholipase Cy1, SRC, PI3K and STAT3. Gab1 can indirectly bind to MET via Grb2. Those adapter molecules allow PI3K-AKT, STAT3, Ras-MAPK, and NF-kB downstream pathways to be activated by HGF/induced MET, promoting cancer cell survival, proliferation, differentiation, motility, invasion, migration, and survival [34,39,40,95,97].
Pleiotropic effects of the MET oncogene include command and signaling of membrane proteins and other RTKs, including EGFR. It is known that other biomarkers mutations in NSCLC, like EGFR-activating mutations in combination with HGF overexpression, can cause upregulation of MET phosphorylation, amplification of PI3K-AKT-mTOR and RAS-MAPK signaling pathway leading to tumor treatment resistance against from multiple drugs, including Gefitinib and Erlotinib. This acquired resistance is mainly observed in c-MET (MET family member) but is not fully understood [39,96,98].
Moreover, MET-mediated signaling gene amplification and protein overexpression can be related to METex14 mutations - most commonly exon 14 skipping in slicing - and may coexist with MET gene copies multiplication causing NSCLC positive EGFR driver mutations resistance. The regulatory portion of exon 14 in the MET gene DNA is responsible for directing the gene's correct transcriptional signaling. In its regularizing cascade, after being connected to its ligand, the overactivation control of MET is avoided through its endocytosis, from which MET can be degraded and recycled. CBL (E3 ubiquitin ligase casitas B-lineage lymphoma) recognizes the juxtamembrane phosphorylated tyrosine residual Tyr1003 - encoded by METex14 - and recruits endocytosis regulators so that MET is ubiquitinated and degraded. Therefore, when there is the complete loss of segment 14, point mutations or insertions (indels) that mimic it, exons 13 and 15 are fused and the juxtamembrane region encoded is loss (as well as Tyr1003 domain) in such a way as to deregulate MET transcription and, consequently, lead to its over-signaling and increased stability (without correct degradation) and exacerbated tumor growth [38-40]. Beyond that, it is known that preferential merging genes with MET, leading to fusion proteins - like TPR-MET in lung cancer - also disrupt the original pathway, but the Exon 14´s insertion into TPR-MET may reduce its oncogenic potential.
ROS1: The ROS1 gene is located at chromosome 6 (6q22.1) and encodes the transmembrane RTK namesake. [54] This receptor has an extracellular ligand-binding domain (which has not yet had its function well established), a transmembrane domain, and an intracellular TK domain [99]. As discussed, gene fusion, overexpression, or mutations can increase ROS1 kinase activities similar to the ALK fusion protein [44,45]. Thus, as a constitutively activated RTK, ROS1 can induce oncogenic cell survival and proliferation. This fact is due to the ability of ROS1 autophosphorylation to activate different downstream molecules, such as i) AKT-mTOR, RAS-MAPK; ii) guanine nucleotide exchange factor 1 (VAV3); iii) Src homology 2 (SH2) domain-containing phosphatase (SHP1/2) NH2-terminal domains and a C-terminal protein-tyrosine phosphatase domains (SHP-1 and SHP-2); iv) cytoskeleton and cell-to-cell interaction proteins pathways (B1-integrin and B1-catenin). As stated, these molecules are responsible for controlling cell proliferation, transformation, and early and late apoptosis programs, similar to ALK fusion protein [99,100].
The role of ROS1 in NSCLC crops up by fusion between gene partners (most frequently CD74, but also SLC34A2, EZR, TMP3, and SDC4) and ROS1 kinase domains, which boosts the kinase activity role to generate oncogenic protein expression [44,49]. Hence, activated ROS1 kinase promotes increased cell proliferation, survival, and migration due to JAK-STAT, PI3K-AKT, and MAPK upregulation [44]. In diagnosed NSCLC with ROS-CD74 fusion and that are Crizotinib-sensitive, further mutations can take place, such as ROS-D2033N (aspartic acid to asparagine substitution within ATP binding-site) and L2155S (hindrance of drug binding) variant mutations [44] (Figure 2).
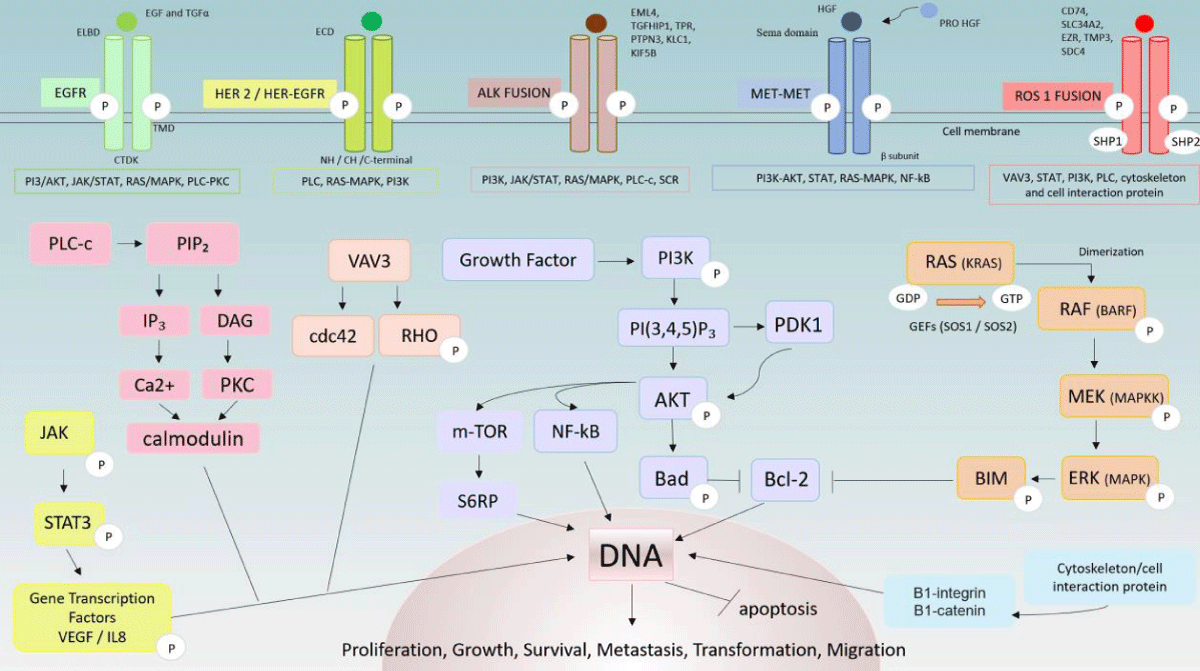
Figure 2: Summary of GFR, HER2, ALK, MET, and ROS1 downstream signaling JAK-STAT, IP3, PI3-AKT, MEK-ERK pathways that lead to cell proliferation, growth, metastasis, transformation, migration, and anti-apoptosis. EGFR binds to EGF and TGFα in the extracellular ELBD domain. HER 2 may fuse with another HER2 or EGFR when activated by its ligands in the ECD domain. ALK fusions are induced by EML4, TGFHIP1, TPR, PTPN3 KLC1, and KIF5B ligands, and MET fusions are activated by HGF (originally inactivated pro-HGF) binding to the Sema domain. ROS1 partners are predominantly CD74, SLC34A2, EZR, TMP3 and SDC4. KRAS's correct function depends on GDP conversion to GTP induced by GEFs. Dimerization and phosphorylation of intracellular domains lead to activation of downstream signaling pathways PLC-c/PKC, JAK-STAT, RAS-MAPK and PI3K AKT responsible for cell migration, proliferation, growth, survival, and transformation [4-6,34,38-40,56-58,65-67,84,88,90,94].
BRAF: BRAF is an essential step in the MAPK-ERK cascade activation, which plays a major role in controlling cell growth and regulation [6]. As mentioned, BRAF mutations enhance the intracellular signal transmission upon EGFR activation. For ERK to be activated, RAF needs to be firstly dimerized by RAS, a process that is limited by a feedback mechanism [54,101]. Specifically, in normal conditions, the activation and phosphorylation of EGFR anchors an adaptor protein which recruits RAS next activating MEK and ERK via RAF [102]. However, mutant BRAF evades feedback inhibition (despite insufficient RAS activity), since BRAF-V600 and non-V600 are RAS-independent proteins and can still be activated [101]. Since ERK phosphorylates multiple kinases, such as nuclear transcription factors, the resulting changes in the RAF protein activity and consequently in gene expression, promote changes in cellular behavior [102].
In abnormal oncogenic tumors, interruption of the operation for negative feedback added to the amplification of the pathway signaling due to BRAF mutations, results in its behavior as oncogenic drivers, which can lead to unchecked cell growth and proliferation. Lastly, the PI3K-AKT parallel pathway is intrinsically linked to the MAPK pathway controlled by RAS, since both ERK and AKT indirectly inhibit the disconnection of bcl-2 from BAX and BAK. If that were to occur, it would induce cell apoptosis. Therefore, this bypass signaling provides the tumor with an alternative pathway for malignant progression [84,102-105].
KRAS: KRAS and its homologs NRAS and HRAS are members of the RAS-like GTPase protein family. It is located internally on the surface of the cell membrane [56,57] and encodes two variants: KRAS 4A (modulator of glucose metabolism, highly expressed in tumors) and KRAS4B (dominant form) [57,58]. Like RAS in normal tissues, KRAS in its inactive state (KRAS-GDP) relies on extracellular stimulation from growth factors such as guanine nucleotide exchange factors (GEFs) (such as, for example, SOS1) and GTPase-activating proteins (GAPs) to exchange GDP for GTP and thus assume its active conformation (KRAS-GTP). SOS1 can also be recruited to the vicinity of the membrane by activation of growth factors from other RTKs. With this proximity, it enables GTP binding to KRAS, forming KRAS in its active conformation. When functioning, KRAS-GTP associates with other effector proteins (such as MAPK, directly activating RAF and indirectly activating ERK kinase activity, and PI3K), stimulating the transmission of signal cascades for cell proliferation, survival, and differentiation. GTPase-activating proteins (GAPs) increase GTPase activity, converting the protein back to its inactive form of KRAS-GDP [56,58,84].
However, missense mutations in KRAS decrease the intrinsic GTPase activity of GAP and keep it in its active state constitutively bound to GTP, triggering intracellular signals independently [56,57]. Therefore, tumorigenesis mediated by mutated KRAS arises from enhanced cellular proliferation potential, survival, stromal inflammation, and fatty acid synthesis [56,58,106].
Among the co-mutations associated with KRAS, the Serine/Threonine Kinase 11 (STK11) gene encodes Liver kinase B1 (LKB1), a protein that phosphorylates AMPK (adenosine monophosphate-activated protein kinase) and related kinases. AMPK phosphorylates programmed cell death 1 ligand 1 (PDL-1), a ligand of programmed cell death protein-1 (PD-1). The binding of PDL-1 to PD1 in Cytotoxic T lymphocytes (CTLs) switches off the anti-tumoral activity of these cells. Phosphorylated PDL1 is ubiquitinated and degraded by proteasomes, thus not blocking CTLs and allowing tumor combat. STK11 inactivation prevents the phosphorylation and degradation of PDL-1, increasing its binding to PD1. Therefore, the tumor immune microenvironment is potentiated, excluding T-cell action and showing innate resistance to PD-L1 inhibition [56,84].
KEAP1 is an adaptor protein (ligase) that connects to CRL3 and Nrf2. Thus, under basal conditions, Nrf2 is degraded by the ubiquitin-proteasome system in the cytosol. Under stress conditions, the KEAP1-NRF2 interaction is disrupted, and free NRF2 translocates to the nucleus and acts as a transcription factor for antioxidant proteins. PDL1 is a direct transcriptional factor of NFR2. Therefore, inactivating mutations in the KEAP1 gene also result in an altered tumor immune microenvironment where free NFR2 targeting PDL1 increases T-cell inactivity [56] and has been shown as an independent factor for primary resistance to monotherapy with PD-1/PD-L1 inhibitors in lung adenocarcinoma with KRAS gene mutation and immunotherapy [56,57].
Moreover, newly produced water-soluble KRAS proteins need to go through prenylation, a post-translational modification, so that they can bind to the membrane (lipophilic). This process is mainly commanded by the farnesyltransferase enzyme (and alternatively by geranylgeranyl transferase I, when farnesyltransferase is inhibited), but the attempt to block this step causes toxic side effects [58].
HER3: One of the main correlations of HER 3 with tumorigenesis relies on its overactivation mediated by Neuromodulin 1. HER3 is a pseudo kinase that has an inactive kinase domain that may pair with other ErbB members. In turn, Neuromodulin 1 (NRG1) - also known as ARIA, HRG, GGF, NDF - oncogene expressed in solid tumors (as well as in 0,2% of lung mutated cells) and its four isoforms (NRG1 type I to IV) encodes an eponymous chimeric protein - a member of the Epidermal Growth Factor (EGF) ligand family. NRG1´s structure is divided into an intracellular segment, a transmembrane portion, and an extracellular region (formed by an EGF-like domain that binds to an immunoglobulin through a glyco-amino acid) [65,107,108].
NRG1 binds to ERbB family receptors (mainly HER3 and 4), triggering asymmetric heterodimerization (as an example, HER2-HER3 associated) forming oncogenic dimers, a mechanism that allows ErbB tyrosine residuals phosphorylation in the C-terminal tail segments [65,107]. These intermolecular interactions allow the binding of signals-transmitting molecules so that the PI3K pathway becomes overactivated in tumorigenesis [107]. This fusion mechanism is associated with poor overall survival in lung cancer.
Other upstream fusion partners (CD74 31%, SLC33A2 16%, ATP1B1 13%, SDC4 9%, RBPMS 4%, CDH1 and VTCN1 both 2%) may continuously activate downstream signaling pathways and have been shown in multiple invasive mucinous adenocarcinomas of the lung, a histological type of lung cancer associated with KRAS [79,108,109]. Clinical reports have demonstrated that NRG1-SLC3A2 fusions were identified - concomitantly with CD74-NRG1, KRAS, NRAS Q61L, and AML4-ALK fusions. SLC3A2-NRG1 resultant proteins have a SLC3A2 transmembrane domain and an intracellular domain analogous to NRG1 - NRG1 type III - linked to an EGF-like domain. This resulting configuration is essential for PI3K/AKT/TOR pathway hyperactivation, promoting cell migration, proliferation, and tumoral growth starting from dimerization and phosphorylation of ErbB2-ErbB3 induced by NRG1 [109]. As a result of a somatic genomic event, CD74-NRG1 fusions can be found in ~27% of never-smoker women [109] and also lead to the externalization of the NRG1 III EGF-like domain as a potential ligand for dimerized HER receptors. Moreover, a clinical study demonstrated an occurrence of 60% of KRAS and CD74 fusion in the mentioned histological subtype [110].
NTRK: The mature TRKA, B, or C receptors of the tropomyosin family are the result of post-translational glycosylation modifications of the extracellular domain of their precursor proteins, the TRKs. In general, these receptors have similar compositions: an intracellular domain with variable length, a transmembrane region, and an extracellular domain, composed of two cysteine clusters (C1 and C2) interconnected by three Leucine-rich repeat proteins. The C2 region is connected to the cell membrane through two immunoglobulin ligands (Ig1 and Ig2). TRK ligands connect to Ig2 and other extracellular domains of each receptor and are, preferably, specific neurotrophic factors: the nerve growth factor (NGF) binds to TRKA present in the nervous system; brain-derived neurotrophic factor (BDNF) and neutrophil 4 (NT-4) to TRKB and neurotrophin 3 (NT-3) to TRKC). It is through these connections that the transmission of intracellular signals is possible. Erroneous signal transduction in this cascade can occur by the alternative linkage - chromosomal translocation - of NTRK to different genes, such as ETV6, LMNA, MPRIP, or TPM3. As a result, NRTK fusions form TRKA or B or C protein receptors that are constitutively activated and indiscriminately propagate hyperproliferation and growth and cellular signals [5,66,67].
NGF or BDNF binds to the immunoglobulin domain of TRKA and TRKB respectively, leading to dimerization. This step is important to promote both activation and autophosphorylation of the tyrosine kinase internal and juxtamembrane domains and the recruitment of specific tyrosines. Once phosphorylated, TK domains are docking sites to SHC - a common and PTB adaptor protein - and PLCy proteins [111]. Studies evaluating the expression of neurotrophins and their protein receptors in cancer patients have observed the connection of BDNF and TRKB in the growth and metastasis of NSCLC tumors and NGF factors linked to TRKA receptors mainly in adenocarcinomas and squamous cells [112,113]. Phosphorylated Y490 binds to SHC (homologous and collagen-like protein) - a common forerunner of the Ras/MAPK pathway and the PI3K/Akt pathways - and to FRS2 (Fibroblast Growth Factor Receptor Substrate 2) that recruits GRB2, CRK, and SRC correlated to MAPK; 7785 binds to PLCy proteins, increasing Ca2+ levels and PKC activation. Thus, different cascades with remarkable and possible interaction and crosstalk are activated [114].
RET: The RET RTK has an extracellular N-terminal ligand-binding region with four cadherin domains and a cysteine-rich domain connected to an intracellular C-terminal TK domain and its specific isoform tails. 9, 43 and 51 C-terminal amino acids form three different RET isoforms with distinguishing carboxy-terminal amino acids, because of alternative splicing: RET9 (higher expressed), RET43 (lower expressed), and RET51 respectively. The ligand-induced heterodimer conformation allows this complex to autophosphorylation of the TKIs regions which serve as docking sites to adaptor proteins, a first step to PI3K/AKT, JAK/STAT, and RAS/MAPK pathways downstream activation [76,77,79].
Unlike other tyrosine kinase receptors, RET receptors depend on two connections to be activated: with their ligands and their respective cofactors that form a binary complex that recruits RET. Primary ligands GDNF, neurturin (NRTN), artemin (ARTN), persephin (PSPN), and GDF15 are GFLs (glial cell line-derived neurotrophic factor (GDNF) family ligands) and the first four ones are members of transforming growth factor beta (TGFb) superfamily. All cofactors are members of the GFRAα - growth factor receptor-alfa - family. GDNF ligand binds to the cofactor GFRAα1; NRTN binds to GFRAα2; ARTN to GFRAα3 and PSPN to GFRAα4; GDF15 binds to GFRAL. These findings are important for the intracellular autophosphorylation domains Y687, Y752, Y806, Y809, Y826, Y900, Y905, Y928, Y981, Y1015, and Y1062 to be activated and become sites of adapter protein coupling. Y1062 binds to Enigma, Dok1/4/5, Shc, IRS1/2 and FRS2 proteins, activating the previously mentioned pathways [77-79,81]. It is observed that when fusion genes (mainly, KIF5B and CCDC6) fuse with RET, the resulting proteins formed can generate homodimerization, autophosphorylation, and, consequently, RET activation independent of its ligands.
It is important to note that understanding the different intracellular signaling pathways and how they interrelate enables the development of therapies that combine drugs targeting different molecules within these pathways. Combined targeted therapies (such as the combination of EGFR inhibitors with MET or HER2 inhibitors) allow for the exploration of new therapeutic strategies against tumor resistance mechanisms that develop both during and after treatment. Additionally, the combined blockade of downstream pathways also enhances the efficiency of therapies against molecular targets that are still newly explored (such as KRAS G12C therapies). Finally, the development of next-generation inhibitors and the validation of combining well-established therapies (such as chemotherapy) with new inhibitors also enable the overcoming of tumor resistance (Figure 3).
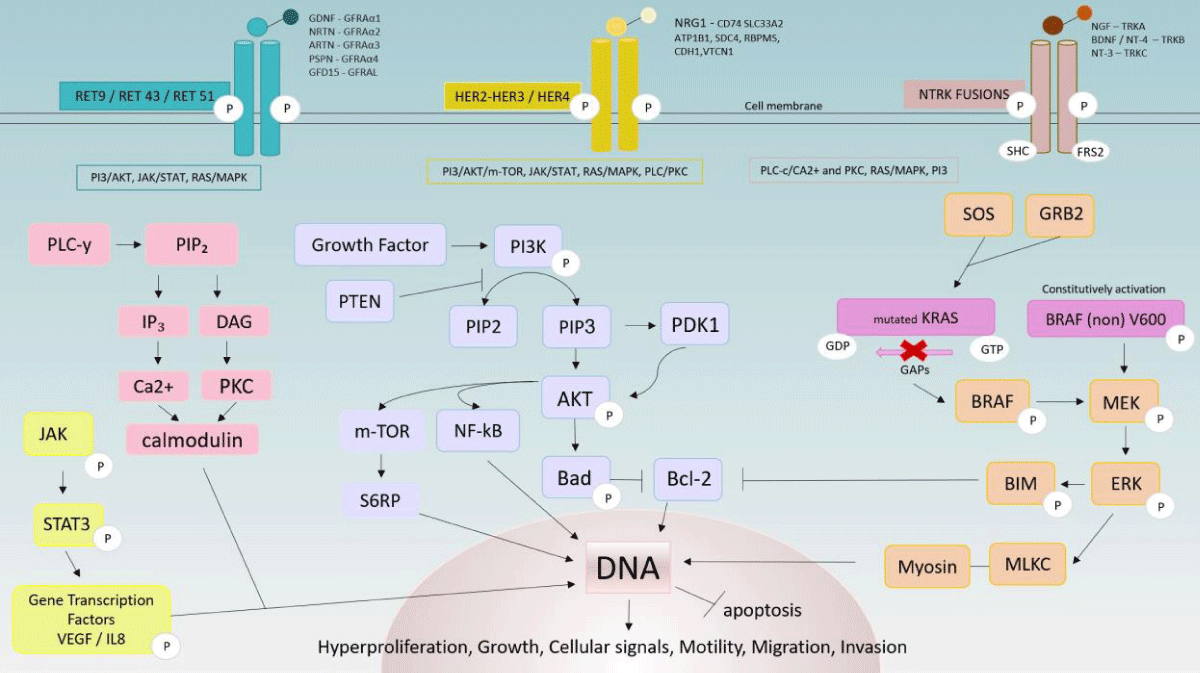
Figure 3: Summary of RET, HER3/4, NTRK, mutated KRAS and BRAF V600 and non-V600. Ret and its isoforms are activated by a double connection with both RET ligands (GFLs) and cofactors (GFRAα family). When activated, its intracellular domains are responsible for conducting signals via adapter proteins, such as Shc and FRS2. NRG1 plays an important role in the HER3 pathway. NRG1 binds to the ERbB family or upstream fusion partners triggering phosphorylation and formation of oncogenic dimers that have intracellular binding docking sites for signal-transmitting molecules. Each TRK ligand connects, preferably, to specific neurotrophic factors, leading to phosphorylation and activation of tyrosine residuals. TK domains are docking sites to SHC, FRS2, and PLCy, molecules part of different signaling pathways. SOS molecules have homologues - 1 and 2 - that catalyze the substitution of GDP for GTP, to activate GTP-dependent KRAS. The opposite conversion - which inhibits KRAS - is done by GAPs. Mutated KRAS, however, is capable of evading the action of GAPs, remaining active with GTP and, therefore, enhancing its signaling cascade. BRAF mutated forms are capable of being self-activated independently of their dimerization by RAS so that the remainder of the MEK/ERK cascade becomes overactivated. These mechanisms lead to the activation of downstream signaling pathways PLC-c/PKC, JAK-STAT, RAS-MAPK, and PI3K AKT responsible for cell migration, proliferation, growth, survival, and transformation [4-5,54,56-59,65-67,76-81,84,101-114].
Globally, 40% of cancer-related deaths are the result of NSCLC. The knowledge about biomarkers and their molecular mechanisms plays an important role in the development of efficient target therapies against mutated cell resistance (proliferation, growth, migration, metastasis, and survival).
Considering the evolution of biomarkers, molecular pathways, and gene research, many of which are still on track testing combinations and interaction between the multiple therapy drugs, we can speculate that the development of new therapies and the improvement of specifically mentioned drugs, mainly EGFR-target therapies, may also provide combined treatment against mutant receptors HER2, HER3, MET, and ROS1. Furthermore, BRAF mutation studies may indirectly reduce the development of tumoral resistance against membrane-receptors-targeted therapies; and KRAS directly targeted drugs (ARS-853) may be developed as an alternative treatment for indirect inhibitors.
The ongoing exploration of novel combination regimens offers a promising avenue to overcome resistance in NSCLC. Simultaneously targeting multiple pathways reduces the potential emergence of resistance, enhancing therapeutic efficacy and improving outcomes. Future research should focus on identifying the combined molecular mechanisms that predispose to resistance and determining the most effective therapeutic combinations to combat them, based on the genetic and molecular tumor profile. In this context, prevalent biomarkers play a crucial role in guiding therapy.
Practice points
- Worldwide, cancer death occurs, primarily, due to lung cancer, mainly as a result of NSCLC.
- Mostly prevalent in NSCLC adenocarcinoma, EGFR-activating mutations have osimertinib treatment as the consolidated standard therapy. However, the development of resistance after the use of third and fourth-generation TKIs and salvage chemotherapy underscores the need for alternative approaches, such as the combined use of osimertinib and chemotherapy or chemotherapy and amivantamab.
- Advanced studies of therapies for NSCLC have included Entrectinib and Larotrectinib, both first-generation tropomyosin receptor kinase (TRK) inhibitors in NTRK (NTRK1, NTRK2, NTRK3) mutation-positive patients.
- The EML4-ALK fusion protein is primarily targeted by Alectinib, Brigatinib, and Lorlatinib, particularly in Crizotinib-resistant patients, with the latter exhibiting high CNS penetration.
- Crizotinib has also been established as the FDA's first-choice standard drug for therapy of ROS1-positive mutations, along with Entrectinib. Another drug designed for ROS1-positive mutations and overcoming resistance in disease progression is Lorlatinib.
- Knowledge about mutations associated with exon 14 becomes important for understanding the disorder of the MET regulatory pathway. Drugs acting on this mutation include Capmatinib, Tepotinib, and Crizotinib.
- RET fusions are targeted by selectively TKI inhibitors: Selpercatinib and Pralsetinib
- The treatment of HER-2-positive cancers conventionally involves the use of pan-HER inhibitors. However, therapeutic enhancement proposes the use of Trastuzumab antibody therapy. Two possible associations with this medication have shown response to developed resistance: TDM1, conjugated with the cytotoxic agent (emtansine - DM1), and TDX-d (linked with topoisomerase I inhibitor -Deruxtecan DX-d).
- The HER3 pathway activated by NRG1 is a target therapy with phase I and II studies being developed. So far, Zenocutuzumab has shown positive indications as a targeted therapy for HER3.
- The 50% prevalence of the BRAF-600 mutation in NSCLC can be treated with either Trametinib and Dabrafenib mono or combined therapy.
- Treatment KRAS mutations target therapies focus on KRAS-G12C inhibitors and indirect inhibition.
Author contributions
Conceptualization, I.S.; writing—original draft preparation, I.S., F.M.S, and J.R.B.; writing—review and editing, I.S., J.R.B.; supervision, F.M.S., and J.R.B.; All authors have read and agreed to the published version of the manuscript.
Data availability statement: The data can be shared upon request.
I would like to thank my family for all the support during my research and writing and my professors at Faculdade Israelita de Ciências da Saúde Albert Einstein for providing me with prior contact with the academic world, which was essential to making research viable, and my supervisors, Dr. Fernando Moura and Dr. Juliana Beal, for the opportunity to develop this article. The figures were made on PowerPoint.
- Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008 May;83(5):584-94. doi: 10.4065/83.5.584. PMID: 18452692; PMCID: PMC2718421.
- Zheng M. Classification and Pathology of Lung Cancer. Surg Oncol Clin N Am. 2016 Jul;25(3):447-68. doi: 10.1016/j.soc.2016.02.003. PMID: 27261908.
- Vyse S, Huang PH. Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer. Signal Transduct Target Ther. 2019 Mar 8;4:5. doi: 10.1038/s41392-019-0038-9. PMID: 30854234; PMCID: PMC6405763.
- Haratake N, Seto T. NTRK fusion-positive non-small-cell lung cancer -The diagnosis and targeted therapy-. Clinical Lung Cancer. 2020 Oct;22(1).
- Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017 Oct 25;17(11):637-658. doi: 10.1038/nrc.2017.84. PMID: 29068003.
- Aoki MN, Amarante MK, de Oliveira CEC, Watanabe MAE. Biomarkers in Non-Small Cell Lung Cancer: Perspectives of Individualized Targeted Therapy. Anticancer Agents Med Chem. 2018;18(15):2070-2077. doi: 10.2174/1871520618666180827102101. PMID: 30147015.
- Nan X, Xie C, Yu X, Liu J. EGFR TKI as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer. Oncotarget. 2017 Aug 9;8(43):75712-75726. doi: 10.18632/oncotarget.20095. PMID: 29088904; PMCID: PMC5650459.
- Harrison PT, Vyse S, Huang PH. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin Cancer Biol. 2020 Apr;61:167-179. doi: 10.1016/j.semcancer.2019.09.015. Epub 2019 Sep 25. PMID: 31562956; PMCID: PMC7083237.
- Lavacchi D, Mazzoni F, Giaccone G. Clinical evaluation of dacomitinib for the treatment of metastatic non-small cell lung cancer (NSCLC): current perspectives. Drug Des Devel Ther. 2019 Sep 6;13:3187-3198. doi: 10.2147/DDDT.S194231. PMID: 31564835; PMCID: PMC6735534.
- Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T, Okamoto I, Zhou C, Cho BC, Cheng Y, Cho EK, Voon PJ, Planchard D, Su WC, Gray JE, Lee SM, Hodge R, Marotti M, Rukazenkov Y, Ramalingam SS; FLAURA Investigators. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med. 2018 Jan 11;378(2):113-125. doi: 10.1056/NEJMoa1713137. Epub 2017 Nov 18. PMID: 29151359.
- Passaro A, Wang J, Wang Y, Lee SH, Melosky B, Shih JY, Wang J, Azuma K, Juan-Vidal O, Cobo M, Felip E, Girard N, Cortot AB, Califano R, Cappuzzo F, Owen S, Popat S, Tan JL, Salinas J, Tomasini P, Gentzler RD, William WN Jr, Reckamp KL, Takahashi T, Ganguly S, Kowalski DM, Bearz A, MacKean M, Barala P, Bourla AB, Girvin A, Greger J, Millington D, Withelder M, Xie J, Sun T, Shah S, Diorio B, Knoblauch RE, Bauml JM, Campelo RG, Cho BC; MARIPOSA-2 Investigators. Amivantamab plus chemotherapy with and without lazertinib in EGFR-mutant advanced NSCLC after disease progression on osimertinib: primary results from the phase III MARIPOSA-2 study. Ann Oncol. 2024 Jan;35(1):77-90. doi: 10.1016/j.annonc.2023.10.117. Epub 2023 Oct 23. PMID: 37879444.
- Cho BC, Kim DW, Spira AI, Gomez JE, Haura EB, Kim SW, Sanborn RE, Cho EK, Lee KH, Minchom A, Lee JS, Han JY, Nagasaka M, Sabari JK, Ou SI, Lorenzini P, Bauml JM, Curtin JC, Roshak A, Gao G, Xie J, Thayu M, Knoblauch RE, Park K. Amivantamab plus lazertinib in osimertinib-relapsed EGFR-mutant advanced non-small cell lung cancer: a phase 1 trial. Nat Med. 2023 Oct;29(10):2577-2585. doi: 10.1038/s41591-023-02554-7. Epub 2023 Sep 14. PMID: 37710001; PMCID: PMC10579096.
- Park K, Haura EB, Leighl NB, Mitchell P, Shu CA, Girard N, Viteri S, Han JY, Kim SW, Lee CK, Sabari JK, Spira AI, Yang TY, Kim DW, Lee KH, Sanborn RE, Trigo J, Goto K, Lee JS, Yang JC, Govindan R, Bauml JM, Garrido P, Krebs MG, Reckamp KL, Xie J, Curtin JC, Haddish-Berhane N, Roshak A, Millington D, Lorenzini P, Thayu M, Knoblauch RE, Cho BC. Amivantamab in EGFR Exon 20 Insertion-Mutated Non-Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results From the CHRYSALIS Phase I Study. J Clin Oncol. 2021 Oct 20;39(30):3391-3402. doi: 10.1200/JCO.21.00662. Epub 2021 Aug 2. PMID: 34339292; PMCID: PMC8791812.
- Shu CA, Goto K, Cho BC, Griesinger F, Yang JCH, Felip E, et al. CHRYSALIS-2: A phase 1/1b study of lazertinib as monotherapy and in combination with amivantamab in patients with EGFR-mutant NSCLC. Journal of Clinical Oncology. 2021 May 20;39(15_suppl):TPS9132–2.
- Zhou C, Tang KJ, Cho BC, Liu B, Paz-Ares L, Cheng S, Kitazono S, Thiagarajan M, Goldman JW, Sabari JK, Sanborn RE, Mansfield AS, Hung JY, Boyer M, Popat S, Mourão Dias J, Felip E, Majem M, Gumus M, Kim SW, Ono A, Xie J, Bhattacharya A, Agrawal T, Shreeve SM, Knoblauch RE, Park K, Girard N; PAPILLON Investigators. Amivantamab plus Chemotherapy in NSCLC with EGFR Exon 20 Insertions. N Engl J Med. 2023 Nov 30;389(22):2039-2051. doi: 10.1056/NEJMoa2306441. Epub 2023 Oct 21. PMID: 37870976.
- Anna Rachel Minchom, Krebs MG, Byoung Chul Cho, Lee SH, Leighl NB, O’Neil B, et al. Subcutaneous amivantamab (ami) in patients (pts) with advanced solid malignancies: The PALOMA study—Updated safety and identification of the recommended phase 2 dose. Journal of Clinical Oncology. 2023 Jun 1;41(16_suppl):9126–6.
- Chouaid C, Bosquet L, Girard N, Kron A, Scheffler M, Griesinger F, Sebastian M, Trigo J, Viteri S, Knott C, Rodrigues B, Rahhali N, Cabrieto J, Diels J, Perualila NJ, Schioppa CA, Sermon J, Toueg R, Erdmann N, Mielke J, Nematian-Samani M, Martin-Fernandez C, Pfaira I, Li T, Mahadevia P, Wolf J. An Adjusted Treatment Comparison Comparing Amivantamab Versus Real-World Clinical Practice in Europe and the United States for Patients with Advanced Non-Small Cell Lung Cancer with Activating Epidermal Growth Factor Receptor Exon 20 Insertion Mutations. Adv Ther. 2023 Mar;40(3):1187-1203. doi: 10.1007/s12325-022-02408-7. Epub 2023 Jan 18. PMID: 36652175; PMCID: PMC9988783.
- Haikala HM, Lopez T, Köhler J, Eser PO, Xu M, Zeng Q, Teceno TJ, Ngo K, Zhao Y, Ivanova EV, Bertram AA, Leeper BA, Chambers ES, Adeni AE, Taus LJ, Kuraguchi M, Kirschmeier PT, Yu C, Shiose Y, Kamai Y, Qiu Y, Paweletz CP, Gokhale PC, Jänne PA. EGFR Inhibition Enhances the Cellular Uptake and Antitumor-Activity of the HER3 Antibody-Drug Conjugate HER3-DXd. Cancer Res. 2022 Jan 1;82(1):130-141. doi: 10.1158/0008-5472.CAN-21-2426. Epub 2021 Sep 21. PMID: 34548332; PMCID: PMC8732289.
- Mazières J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, Besse B, Blons H, Mansuet-Lupo A, Urban T, Moro-Sibilot D, Dansin E, Chouaid C, Wislez M, Diebold J, Felip E, Rouquette I, Milia JD, Gautschi O. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 2013 Jun 1;31(16):1997-2003. doi: 10.1200/JCO.2012.45.6095. Epub 2013 Apr 22. PMID: 23610105.
- Iqbal N, Iqbal N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol Biol Int. 2014;2014:852748. doi: 10.1155/2014/852748. Epub 2014 Sep 7. PMID: 25276427; PMCID: PMC4170925.
- Pillai RN, Behera M, Berry LD, Rossi MR, Kris MG, Johnson BE, Bunn PA, Ramalingam SS, Khuri FR. HER2 mutations in lung adenocarcinomas: A report from the Lung Cancer Mutation Consortium. Cancer. 2017 Nov 1;123(21):4099-4105. doi: 10.1002/cncr.30869. Epub 2017 Jul 25. PMID: 28743157; PMCID: PMC5650517.
- Dziadziuszko R, Smit EF, Dafni U, Wolf J, Wasąg B, Biernat W, Finn SP, Kammler R, Tsourti Z, Rabaglio M, Ruepp B, Roschitzki-Voser H, Stahel RA, Felip E, Peters S. Afatinib in NSCLC With HER2 Mutations: Results of the Prospective, Open-Label Phase II NICHE Trial of European Thoracic Oncology Platform (ETOP). J Thorac Oncol. 2019 Jun;14(6):1086-1094. doi: 10.1016/j.jtho.2019.02.017. Epub 2019 Feb 27. PMID: 30825613.
- Li BT, Smit EF, Goto Y, Nakagawa K, Udagawa H, Mazières J, Nagasaka M, Bazhenova L, Saltos AN, Felip E, Pacheco JM, Pérol M, Paz-Ares L, Saxena K, Shiga R, Cheng Y, Acharyya S, Vitazka P, Shahidi J, Planchard D, Jänne PA; DESTINY-Lung01 Trial Investigators. Trastuzumab Deruxtecan in HER2-Mutant Non-Small-Cell Lung Cancer. N Engl J Med. 2022 Jan 20;386(3):241-251. doi: 10.1056/NEJMoa2112431. Epub 2021 Sep 18. PMID: 34534430; PMCID: PMC9066448.
- Li BT, Shen R, Buonocore D, Olah ZT, Ni A, Ginsberg MS, Ulaner GA, Offin M, Feldman D, Hembrough T, Cecchi F, Schwartz S, Pavlakis N, Clarke S, Won HH, Brzostowski EB, Riely GJ, Solit DB, Hyman DM, Drilon A, Rudin CM, Berger MF, Baselga J, Scaltriti M, Arcila ME, Kris MG. Ado-Trastuzumab Emtansine for Patients With HER2-Mutant Lung Cancers: Results From a Phase II Basket Trial. J Clin Oncol. 2018 Aug 20;36(24):2532-2537. doi: 10.1200/JCO.2018.77.9777. Epub 2018 Jul 10. Erratum in: J Clin Oncol. 2019 Feb 1;37(4):362. doi: 10.1200/JCO.18.02207. PMID: 29989854; PMCID: PMC6366814.
- Peters S, Zimmermann S. Targeted therapy in NSCLC driven by HER2 insertions. Transl Lung Cancer Res. 2014 Apr;3(2):84-8. doi: 10.3978/j.issn.2218-6751.2014.02.06. PMID: 25806285; PMCID: PMC4367663.
- Richard S, Selle F, Lotz JP, Khalil A, Gligorov J, Soares DG. Pertuzumab and trastuzumab: the rationale way to synergy. An Acad Bras Cienc. 2016;88 Suppl 1:565-77. doi: 10.1590/0001-3765201620150178. PMID: 27275646.
- Mar N, Vredenburgh JJ, Wasser JS. Targeting HER2 in the treatment of non-small cell lung cancer. Lung Cancer. 2015 Mar;87(3):220-5. doi: 10.1016/j.lungcan.2014.12.018. Epub 2015 Jan 8. PMID: 25601485.
- Du X, Shao Y, Qin HF, Tai YH, Gao HJ. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac Cancer. 2018 Apr;9(4):423-430. doi: 10.1111/1759-7714.12613. Epub 2018 Feb 28. PMID: 29488330; PMCID: PMC5879058.
- Schrank Z, Chhabra G, Lin L, Iderzorig T, Osude C, Khan N, Kuckovic A, Singh S, Miller RJ, Puri N. Current Molecular-Targeted Therapies in NSCLC and Their Mechanism of Resistance. Cancers (Basel). 2018 Jul 4;10(7):224. doi: 10.3390/cancers10070224. PMID: 29973561; PMCID: PMC6071023.
- Wu W, Haderk F, Bivona TG. Non-Canonical Thinking for Targeting ALK-Fusion Onco-Proteins in Lung Cancer. Cancers (Basel). 2017 Nov 30;9(12):164. doi: 10.3390/cancers9120164. PMID: 29189709; PMCID: PMC5742812.
- Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, Sequist LV, Tan W, Gandhi L, Mino-Kenudson M, Wei GC, Shreeve SM, Ratain MJ, Settleman J, Christensen JG, Haber DA, Wilner K, Salgia R, Shapiro GI, Clark JW, Iafrate AJ. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010 Oct 28;363(18):1693-703. doi: 10.1056/NEJMoa1006448. Erratum in: N Engl J Med. 2011 Feb 10;364(6):588. PMID: 20979469; PMCID: PMC3014291.
- Dagogo-Jack I, Shaw AT. Crizotinib resistance: implications for therapeutic strategies. Ann Oncol. 2016 Sep;27 Suppl 3(Suppl 3):iii42-iii50. doi: 10.1093/annonc/mdw305. PMID: 27573756; PMCID: PMC5003168.
- Gristina V, La Mantia M, Iacono F, Galvano A, Russo A, Bazan V. The Emerging Therapeutic Landscape of ALK Inhibitors in Non-Small Cell Lung Cancer. Pharmaceuticals (Basel). 2020 Dec 18;13(12):474. doi: 10.3390/ph13120474. PMID: 33352844; PMCID: PMC7766858.
- Cecchi F, Rabe DC, Bottaro DP. The Hepatocyte Growth Factor Receptor: Structure, Function and Pharmacological Targeting in Cancer. Curr Signal Transduct Ther. 2011;6(2):146-151. doi: 10.2174/157436211795659955. PMID: 25197268; PMCID: PMC4156285.
- Salgia R. MET in Lung Cancer: Biomarker Selection Based on Scientific Rationale. Mol Cancer Ther. 2017 Apr;16(4):555-565. doi: 10.1158/1535-7163.MCT-16-0472. PMID: 28373408.
- Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J Thorac Oncol. 2017 Jan;12(1):15-26. doi: 10.1016/j.jtho.2016.10.014. Epub 2016 Oct 26. PMID: 27794501; PMCID: PMC5603268.
- Cascone T, Xu L, Lin HY, Liu W, Tran HT, Liu Y, Howells K, Haddad V, Hanrahan E, Nilsson MB, Cortez MA, Giri U, Kadara H, Saigal B, Park YY, Peng W, Lee JS, Ryan AJ, Jüergensmeier JM, Herbst RS, Wang J, Langley RR, Wistuba II, Lee JJ, Heymach JV. The HGF/c-MET Pathway Is a Driver and Biomarker of VEGFR-inhibitor Resistance and Vascular Remodeling in Non-Small Cell Lung Cancer. Clin Cancer Res. 2017 Sep 15;23(18):5489-5501. doi: 10.1158/1078-0432.CCR-16-3216. Epub 2017 May 30. PMID: 28559461; PMCID: PMC5600821.
- Socinski MA, Pennell NA, Davies KD. MET Exon 14 Skipping Mutations in Non-Small-Cell Lung Cancer: An Overview of Biology, Clinical Outcomes, and Testing Considerations. JCO Precis Oncol. 2021 Apr 13;5:PO.20.00516. doi: 10.1200/PO.20.00516. PMID: 34036238; PMCID: PMC8140815.
- Santarpia M, Massafra M, Gebbia V, D'Aquino A, Garipoli C, Altavilla G, Rosell R. A narrative review of MET inhibitors in non-small cell lung cancer with MET exon 14 skipping mutations. Transl Lung Cancer Res. 2021 Mar;10(3):1536-1556. doi: 10.21037/tlcr-20-1113. PMID: 33889528; PMCID: PMC8044480.
- Comoglio PM, Trusolino L, Boccaccio C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat Rev Cancer. 2018 Jun;18(6):341-358. doi: 10.1038/s41568-018-0002-y. PMID: 29674709.
- Takigawa N, Ochi N, Yamane H. Blocking both epidermal growth factor receptor and mesenchymal-to-epithelial transition pathways in EGFR-mutated lung cancer. Translational Lung Cancer Research. 2018 Dec;7(S4):S352–5.
- Liang H, Wang M. MET Oncogene in Non-Small Cell Lung Cancer: Mechanism of MET Dysregulation and Agents Targeting the HGF/c-Met Axis. Onco Targets Ther. 2020 Mar 25;13:2491-2510. doi: 10.2147/OTT.S231257. PMID: 32273721; PMCID: PMC7104217.
- Cheng JT, Yang J, Wu Y. MET second-site mutations in EGFR-mutant, MET-amplified non-small cell lung cancer after resistance to combinatorial targeted therapy. Journal of Clinical Oncology. 2019 May 20;37(15_suppl):e20730–0.
- D'Angelo A, Sobhani N, Chapman R, Bagby S, Bortoletti C, Traversini M, Ferrari K, Voltolini L, Darlow J, Roviello G. Focus on ROS1-Positive Non-Small Cell Lung Cancer (NSCLC): Crizotinib, Resistance Mechanisms and the Newer Generation of Targeted Therapies. Cancers (Basel). 2020 Nov 6;12(11):3293. doi: 10.3390/cancers12113293. PMID: 33172113; PMCID: PMC7694780.
- Al-Sanea MM, Abdelazem AZ, Park BS, Yoo KH, Sim T, Kwon YJ, Lee SH. ROS1 Kinase Inhibitors for Molecular-Targeted Therapies. Curr Med Chem. 2016;23(2):142-60. doi: 10.2174/0929867322666151006093623. PMID: 26438251.
- Kohno T, Nakaoku T, Tsuta K, Tsuchihara K, Matsumoto S, Yoh K, Goto K. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl Lung Cancer Res. 2015 Apr;4(2):156-64. doi: 10.3978/j.issn.2218-6751.2014.11.11. PMID: 25870798; PMCID: PMC4384213.
- Cai W, Li W, Ren S, Zheng L, Li X, Zhou C. Coexistence of three variants involving two different fusion partners of ROS1 including a novel variant of ROS1 fusions in lung adenocarcinoma: a case report. J Thorac Oncol. 2014 Jun;9(6):e43-6. doi: 10.1097/JTO.0000000000000118. PMID: 24828671.
- Patil T, Smith DE, Bunn PA, Aisner DL, Le AT, Hancock M, Purcell WT, Bowles DW, Camidge DR, Doebele RC. The Incidence of Brain Metastases in Stage IV ROS1-Rearranged Non-Small Cell Lung Cancer and Rate of Central Nervous System Progression on Crizotinib. J Thorac Oncol. 2018 Nov;13(11):1717-1726. doi: 10.1016/j.jtho.2018.07.001. Epub 2018 Jul 5. PMID: 29981925; PMCID: PMC6204290.
- Roskoski R Jr. ROS1 protein-tyrosine kinase inhibitors in the treatment of ROS1 fusion protein-driven non-small cell lung cancers. Pharmacol Res. 2017 Jul;121:202-212. doi: 10.1016/j.phrs.2017.04.022. Epub 2017 Apr 30. PMID: 28465216.
- Robinson SD, O'Shaughnessy JA, Cowey CL, Konduri K. BRAF V600E-mutated lung adenocarcinoma with metastases to the brain responding to treatment with vemurafenib. Lung Cancer. 2014 Aug;85(2):326-30. doi: 10.1016/j.lungcan.2014.05.009. Epub 2014 May 21. PMID: 24888229.
- Jiang J, Gao J, Wang G, Lv J, Chen W, Ben J, Wang R. Case Report: Vemurafenib Treatment in Brain Metastases of BRAFS365L -Mutant Lung Papillary Cancer by Genetic Sequencing of Cerebrospinal Fluid Circulating Tumor DNA Detection. Front Oncol. 2021 Jun 11;11:688200. doi: 10.3389/fonc.2021.688200. PMID: 34178685; PMCID: PMC8226071.
- Leonetti A, Facchinetti F, Rossi G, Minari R, Conti A, Friboulet L, Tiseo M, Planchard D. BRAF in non-small cell lung cancer (NSCLC): Pickaxing another brick in the wall. Cancer Treat Rev. 2018 May;66:82-94. doi: 10.1016/j.ctrv.2018.04.006. Epub 2018 Apr 24. PMID: 29729495.
- de Langen AJ, Smit EF. Therapeutic approach to treating patients with BRAF-mutant lung cancer: latest evidence and clinical implications. Ther Adv Med Oncol. 2017 Jan;9(1):46-58. doi: 10.1177/1758834016670555. Epub 2016 Oct 3. PMID: 28203297; PMCID: PMC5298452.
- Piotrowska Z, Sequist LV. Tackling the Next Generation of Resistance in EGFR -Mutant Lung Cancer. Journal of Thoracic Oncology. 2017 Mar;12(3):419–21.
- Marabese M, Ganzinelli M, Garassino MC, Shepherd FA, Piva S, Caiola E, Macerelli M, Bettini A, Lauricella C, Floriani I, Farina G, Longo F, Bonomi L, Fabbri MA, Veronese S, Marsoni S, Broggini M, Rulli E. KRAS mutations affect prognosis of non-small-cell lung cancer patients treated with first-line platinum containing chemotherapy. Oncotarget. 2015 Oct 20;6(32):34014-22. doi: 10.18632/oncotarget.5607. PMID: 26416458; PMCID: PMC4741822.
- Lim TKH, Skoulidis F, Kerr KM, Ahn MJ, Kapp JR, Soares FA, Yatabe Y. KRAS G12C in advanced NSCLC: Prevalence, co-mutations, and testing. Lung Cancer. 2023 Oct;184:107293. doi: 10.1016/j.lungcan.2023.107293. Epub 2023 Jul 13. PMID: 37683526.
- Salgia R, Pharaon R, Mambetsariev I, Nam A, Sattler M. The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC). Cell Rep Med. 2021 Jan 19;2(1):100186. doi: 10.1016/j.xcrm.2020.100186. PMID: 33521700; PMCID: PMC7817862.
- Liu P, Wang Y, Li X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm Sin B. 2019 Sep;9(5):871-879. doi: 10.1016/j.apsb.2019.03.002. Epub 2019 Mar 6. PMID: 31649840; PMCID: PMC6804475.
- Merz V, Gaule M, Zecchetto C, Cavaliere A, Casalino S, Pesoni C, Contarelli S, Sabbadini F, Bertolini M, Mangiameli D, Milella M, Fedele V, Melisi D. Targeting KRAS: The Elephant in the Room of Epithelial Cancers. Front Oncol. 2021 Mar 11;11:638360. doi: 10.3389/fonc.2021.638360. PMID: 33777798; PMCID: PMC7991835.
- Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011 Oct;3(14):1787-808. doi: 10.4155/fmc.11.121. PMID: 22004085; PMCID: PMC3347641.
- Uras IZ, Moll HP, Casanova E. Targeting KRAS Mutant Non-Small-Cell Lung Cancer: Past, Present and Future. Int J Mol Sci. 2020 Jun 17;21(12):4325. doi: 10.3390/ijms21124325. PMID: 32560574; PMCID: PMC7352653.
- Heavey S, Cuffe S, Finn S, Young V, Ryan R, Nicholson S, Leonard N, McVeigh N, Barr M, O'Byrne K, Gately K. In pursuit of synergy: An investigation of the PI3K/mTOR/MEK co-targeted inhibition strategy in NSCLC. Oncotarget. 2016 Nov 29;7(48):79526-79543. doi: 10.18632/oncotarget.12755. PMID: 27765909; PMCID: PMC5346733.
- Scharpenseel H, Hanssen A, Loges S, Mohme M, Bernreuther C, Peine S, Lamszus K, Goy Y, Petersen C, Westphal M, Glatzel M, Riethdorf S, Pantel K, Wikman H. EGFR and HER3 expression in circulating tumor cells and tumor tissue from non-small cell lung cancer patients. Sci Rep. 2019 May 15;9(1):7406. doi: 10.1038/s41598-019-43678-6. PMID: 31092882; PMCID: PMC6520391.
- Jänne PA, Baik C, Su WC, Johnson ML, Hayashi H, Nishio M, Kim DW, Koczywas M, Gold KA, Steuer CE, Murakami H, Yang JC, Kim SW, Vigliotti M, Shi R, Qi Z, Qiu Y, Zhao L, Sternberg D, Yu C, Yu HA. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor-Resistant, EGFR-Mutated Non-Small Cell Lung Cancer. Cancer Discov. 2022 Jan;12(1):74-89. doi: 10.1158/2159-8290.CD-21-0715. Epub 2021 Sep 21. Erratum in: Cancer Discov. 2022 Jun 2;12(6):1598. doi: 10.1158/2159-8290.CD-22-0365. PMID: 34548309; PMCID: PMC9401524.
- Schram AM, Odintsov I, Espinosa-Cotton M, Khodos I, Sisso WJ, Mattar MS, Lui AJW, Vojnic M, Shameem SH, Chauhan T, Torrisi J, Ford J, O'Connor MN, Geuijen CAW, Schackmann RCJ, Lammerts van Bueren JJ, Wasserman E, de Stanchina E, O'Reilly EM, Ladanyi M, Drilon A, Somwar R. Zenocutuzumab, a HER2xHER3 Bispecific Antibody, Is Effective Therapy for Tumors Driven by NRG1 Gene Rearrangements. Cancer Discov. 2022 May 2;12(5):1233-1247. doi: 10.1158/2159-8290.CD-21-1119. PMID: 35135829; PMCID: PMC9394398.
- Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol. 2018 Dec;15(12):731-747. doi: 10.1038/s41571-018-0113-0. PMID: 30333516; PMCID: PMC6419506.
- Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, Mahale S, Davies KD, Aisner DL, Pilling AB, Berge EM, Kim J, Sasaki H, Park S, Kryukov G, Garraway LA, Hammerman PS, Haas J, Andrews SW, Lipson D, Stephens PJ, Miller VA, Varella-Garcia M, Jänne PA, Doebele RC. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med. 2013 Nov;19(11):1469-1472. doi: 10.1038/nm.3352. Epub 2013 Oct 27. PMID: 24162815; PMCID: PMC3823836.
- Drilon A. TRK inhibitors in TRK fusion-positive cancers. Ann Oncol. 2019 Nov;30 Suppl 8:viii23-viii30. doi: 10.1093/annonc/mdz282. Epub 2019 Dec 24. PMID: 32223935.
- Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, Blakely CM, Seto T, Cho BC, Tosi D, Besse B, Chawla SP, Bazhenova L, Krauss JC, Chae YK, Barve M, Garrido-Laguna I, Liu SV, Conkling P, John T, Fakih M, Sigal D, Loong HH, Buchschacher GL Jr, Garrido P, Nieva J, Steuer C, Overbeck TR, Bowles DW, Fox E, Riehl T, Chow-Maneval E, Simmons B, Cui N, Johnson A, Eng S, Wilson TR, Demetri GD; trial investigators. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020 Feb;21(2):271-282. doi: 10.1016/S1470-2045(19)30691-6. Epub 2019 Dec 11. Erratum in: Lancet Oncol. 2020 Feb;21(2):e70. doi: 10.1016/S1470-2045(20)30029-2. Erratum in: Lancet Oncol. 2020 Jul;21(7):e341. doi: 10.1016/S1470-2045(20)30345-4. Erratum in: Lancet Oncol. 2020 Aug;21(8):e372. doi: 10.1016/S1470-2045(20)30382-X. Erratum in: Lancet Oncol. 2021 Oct;22(10):e428. doi: 10.1016/S1470-2045(21)00538-6. PMID: 31838007; PMCID: PMC7461630.
- Kheder ES, Hong DS. Emerging Targeted Therapy for Tumors with NTRK Fusion Proteins. Clin Cancer Res. 2018 Dec 1;24(23):5807-5814. doi: 10.1158/1078-0432.CCR-18-1156. Epub 2018 Jul 9. PMID: 29986850.
- Harada G, Santini FC, Wilhelm C, Drilon A. NTRK fusions in lung cancer: From biology to therapy. Lung Cancer. 2021 Nov;161:108-113. doi: 10.1016/j.lungcan.2021.09.005. Epub 2021 Sep 16. PMID: 34563714; PMCID: PMC8530887.
- Drilon A, Ou SI, Cho BC, Kim DW, Lee J, Lin JJ, Zhu VW, Ahn MJ, Camidge DR, Nguyen J, Zhai D, Deng W, Huang Z, Rogers E, Liu J, Whitten J, Lim JK, Stopatschinskaja S, Hyman DM, Doebele RC, Cui JJ, Shaw AT. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent- Front Mutations. Cancer Discov. 2018 Oct;8(10):1227-1236. doi: 10.1158/2159-8290.CD-18-0484. Epub 2018 Aug 9. PMID: 30093503.
- Hong DS, DuBois SG, Kummar S, Farago AF, Albert CM, Rohrberg KS, van Tilburg CM, Nagasubramanian R, Berlin JD, Federman N, Mascarenhas L, Geoerger B, Dowlati A, Pappo AS, Bielack S, Doz F, McDermott R, Patel JD, Schilder RJ, Tahara M, Pfister SM, Witt O, Ladanyi M, Rudzinski ER, Nanda S, Childs BH, Laetsch TW, Hyman DM, Drilon A. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020 Apr;21(4):531-540. doi: 10.1016/S1470-2045(19)30856-3. Epub 2020 Feb 24. PMID: 32105622; PMCID: PMC7497841.
- Harada G, Drilon A. TRK inhibitor activity and resistance in TRK fusion-positive cancers in adults. Cancer Genet. 2022 Jun;264-265:33-39. doi: 10.1016/j.cancergen.2022.03.002. Epub 2022 Mar 16. PMID: 35334340; PMCID: PMC9133157.
- Papadopoulos KP, Borazanci E, Shaw AT, Katayama R, Shimizu Y, Zhu VW, Sun TY, Wakelee HA, Madison R, Schrock AB, Senaldi G, Nakao N, Hanzawa H, Tachibana M, Isoyama T, Nakamaru K, Deng C, Li M, Fan F, Zhao Q, Gao Y, Seto T, Jänne PA, Ou SI. U.S. Phase I First-in-human Study of Taletrectinib (DS-6051b/AB-106), a ROS1/TRK Inhibitor, in Patients with Advanced Solid Tumors. Clin Cancer Res. 2020 Sep 15;26(18):4785-4794. doi: 10.1158/1078-0432.CCR-20-1630. Epub 2020 Jun 26. PMID: 32591465.
- Cascetta P, Sforza V, Manzo A, Carillio G, Palumbo G, Esposito G, Montanino A, Costanzo R, Sandomenico C, De Cecio R, Piccirillo MC, La Manna C, Totaro G, Muto P, Picone C, Bianco R, Normanno N, Morabito A. RET Inhibitors in Non-Small-Cell Lung Cancer. Cancers (Basel). 2021 Sep 1;13(17):4415. doi: 10.3390/cancers13174415. PMID: 34503226; PMCID: PMC8431193.
- Belli C, Anand S, Gainor JF, Penault-Llorca F, Subbiah V, Drilon A, Andrè F, Curigliano G. Progresses Toward Precision Medicine in RET-altered Solid Tumors. Clin Cancer Res. 2020 Dec 1;26(23):6102-6111. doi: 10.1158/1078-0432.CCR-20-1587. Epub 2020 Jul 14. PMID: 32665298.
- Drilon A, Lin JJ, Filleron T, Ni A, Milia J, Bergagnini I, Hatzoglou V, Velcheti V, Offin M, Li B, Carbone DP, Besse B, Mok T, Awad MM, Wolf J, Owen D, Camidge DR, Riely GJ, Peled N, Kris MG, Mazieres J, Gainor JF, Gautschi O. Frequency of Brain Metastases and Multikinase Inhibitor Outcomes in Patients With RET-Rearranged Lung Cancers. J Thorac Oncol. 2018 Oct;13(10):1595-1601. doi: 10.1016/j.jtho.2018.07.004. Epub 2018 Jul 11. PMID: 30017832; PMCID: PMC6434708.
- Mahato AK, Sidorova YA. RET Receptor Tyrosine Kinase: Role in Neurodegeneration, Obesity, and Cancer. Int J Mol Sci. 2020 Sep 26;21(19):7108. doi: 10.3390/ijms21197108. PMID: 32993133; PMCID: PMC7583994.
- Kato S, Subbiah V, Marchlik E, Elkin SK, Carter JL, Kurzrock R. RET Aberrations in Diverse Cancers: Next-Generation Sequencing of 4,871 Patients. Clin Cancer Res. 2017 Apr 15;23(8):1988-1997. doi: 10.1158/1078-0432.CCR-16-1679. Epub 2016 Sep 28. PMID: 27683183.
- Ferrara R, Auger N, Auclin E, Besse B. Clinical and Translational Implications of RET Rearrangements in Non-Small Cell Lung Cancer. J Thorac Oncol. 2018 Jan;13(1):27-45. doi: 10.1016/j.jtho.2017.10.021. Epub 2017 Nov 8. PMID: 29128428.
- Drilon A, Oxnard GR, Tan DSW, Loong HHF, Johnson M, Gainor J, McCoach CE, Gautschi O, Besse B, Cho BC, Peled N, Weiss J, Kim YJ, Ohe Y, Nishio M, Park K, Patel J, Seto T, Sakamoto T, Rosen E, Shah MH, Barlesi F, Cassier PA, Bazhenova L, De Braud F, Garralda E, Velcheti V, Satouchi M, Ohashi K, Pennell NA, Reckamp KL, Dy GK, Wolf J, Solomon B, Falchook G, Ebata K, Nguyen M, Nair B, Zhu EY, Yang L, Huang X, Olek E, Rothenberg SM, Goto K, Subbiah V. Efficacy of Selpercatinib in RET Fusion-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2020 Aug 27;383(9):813-824. doi: 10.1056/NEJMoa2005653. PMID: 32846060; PMCID: PMC7506467.
- Griesinger F, Curigliano G, Thomas M, Subbiah V, Baik CS, Tan DSW, Lee DH, Misch D, Garralda E, Kim DW, van der Wekken AJ, Gainor JF, Paz-Ares L, Liu SV, Kalemkerian GP, Houvras Y, Bowles DW, Mansfield AS, Lin JJ, Smoljanovic V, Rahman A, Kong S, Zalutskaya A, Louie-Gao M, Boral AL, Mazières J. Safety and efficacy of pralsetinib in RET fusion-positive non-small-cell lung cancer including as first-line therapy: update from the ARROW trial. Ann Oncol. 2022 Nov;33(11):1168-1178. doi: 10.1016/j.annonc.2022.08.002. Epub 2022 Aug 13. PMID: 35973665.
- Alberts B, Bray D, Hopkin K, Johnson A, Lewis J, Raff M, et al. Essential Cell Biology [Internet]. 2016. https://ia800106.us.archive.org/10/items/AlbertsEssentialCellBiology4thEd./Alberts%20-%20Essential%20Cell%20Biology%20%284th%20ed.%29.pdf
- Sundaram MV. RTK/Ras/MAPK signaling. WormBook. 2006 Feb 11:1-19. doi: 10.1895/wormbook.1.80.1. PMID: 18050474; PMCID: PMC4780977.
- Morrison DK. MAP kinase pathways. Cold Spring Harb Perspect Biol. 2012 Nov 1;4(11):a011254. doi: 10.1101/cshperspect.a011254. PMID: 23125017; PMCID: PMC3536342.
- Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012 Sep 1;4(9):a011189. doi: 10.1101/cshperspect.a011189. Erratum in: Cold Spring Harb Perspect Biol. 2015 Apr 01;7(4):a026609. doi: 10.1101/cshperspect.a026609. PMID: 22952397; PMCID: PMC3428770.
- Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci. 2008 May;65(10):1566-84. doi: 10.1007/s00018-008-7440-8. PMID: 18259690; PMCID: PMC3060045.
- Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007 Oct 4;26(45):6469-87. doi: 10.1038/sj.onc.1210477. Epub 2007 Apr 30. PMID: 17471238; PMCID: PMC3021475.
- Lyu H, Han A, Polsdofer E, Liu S, Liu B. Understanding the biology of HER3 receptor as a therapeutic target in human cancer. Acta Pharm Sin B. 2018 Jul;8(4):503-510. doi: 10.1016/j.apsb.2018.05.010. Epub 2018 Jun 2. PMID: 30109175; PMCID: PMC6090011.
- Sabir SR, Yeoh S, Jackson G, Bayliss R. EML4-ALK Variants: Biological and Molecular Properties, and the Implications for Patients. Cancers (Basel). 2017 Sep 5;9(9):118. doi: 10.3390/cancers9090118. PMID: 28872581; PMCID: PMC5615333.
- Bayliss R, Choi J, Fennell DA, Fry AM, Richards MW. Molecular mechanisms that underpin EML4-ALK driven cancers and their response to targeted drugs. Cell Mol Life Sci. 2016 Mar;73(6):1209-24. doi: 10.1007/s00018-015-2117-6. Epub 2016 Jan 11. PMID: 26755435; PMCID: PMC4761370.
- Wong DW, Leung EL, So KK, Tam IY, Sihoe AD, Cheng LC, Ho KK, Au JS, Chung LP, Pik Wong M; University of Hong Kong Lung Cancer Study Group. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009 Apr 15;115(8):1723-33. doi: 10.1002/cncr.24181. PMID: 19170230.
- Takezawa K, Okamoto I, Nishio K, Jänne PA, Nakagawa K. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res. 2011 Apr 15;17(8):2140-8. doi: 10.1158/1078-0432.CCR-10-2798. Epub 2011 Mar 17. PMID: 21415216.
- Hartmann S, Bhola NE, Grandis JR. HGF/Met Signaling in Head and Neck Cancer: Impact on the Tumor Microenvironment. Clin Cancer Res. 2016 Aug 15;22(16):4005-13. doi: 10.1158/1078-0432.CCR-16-0951. Epub 2016 Jul 1. PMID: 27370607; PMCID: PMC6820346.
- Zhang Y, Xia M, Jin K, Wang S, Wei H, Fan C, Wu Y, Li X, Li X, Li G, Zeng Z, Xiong W. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol Cancer. 2018 Feb 19;17(1):45. doi: 10.1186/s12943-018-0796-y. PMID: 29455668; PMCID: PMC5817860.
- Owusu BY, Galemmo R, Janetka J, Klampfer L. Hepatocyte Growth Factor, a Key Tumor-Promoting Factor in the Tumor Microenvironment. Cancers (Basel). 2017 Apr 17;9(4):35. doi: 10.3390/cancers9040035. PMID: 28420162; PMCID: PMC5406710.
- Feng Y, Ma PC. MET targeted therapy for lung cancer: clinical development and future directions. Lung Cancer (Auckl). 2012 Aug 9;3:53-67. doi: 10.2147/LCTT.S23423. PMID: 28210125; PMCID: PMC5312490.
- Gainor JF, Shaw AT. Novel Targets in Non-Small Cell Lung Cancer: ROS1 and RET Fusions. The Oncologist [Internet]. 2013 Jul 1 [cited 2021 Aug 27];18(7):865–75.
- Chin LP, Soo RA, Soong R, Ou SH. Targeting ROS1 with anaplastic lymphoma kinase inhibitors: a promising therapeutic strategy for a newly defined molecular subset of non-small-cell lung cancer. J Thorac Oncol. 2012 Nov;7(11):1625-30. doi: 10.1097/JTO.0b013e31826baf83. PMID: 23070242.
- Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, de Stanchina E, Abdel-Wahab O, Solit DB, Poulikakos PI, Rosen N. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell. 2015 Sep 14;28(3):370-83. doi: 10.1016/j.ccell.2015.08.001. Epub 2015 Sep 3. PMID: 26343582; PMCID: PMC4894664.
- Wee P, Wang Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers (Basel). 2017 May 17;9(5):52. doi: 10.3390/cancers9050052. PMID: 28513565; PMCID: PMC5447962.
- Govender D, Chetty R. Gene of the month: BRAF. J Clin Pathol. 2012 Nov;65(11):986-8. doi: 10.1136/jclinpath-2012-200960. Epub 2012 Sep 3. PMID: 22944621.
- Smalley I, Smalley KSM. ERK Inhibition: A New Front in the War against MAPK Pathway-Driven Cancers? Cancer Discov. 2018 Feb;8(2):140-142. doi: 10.1158/2159-8290.CD-17-1355. PMID: 29431672.
- Papadimitrakopoulou V. Development of PI3K/AKT/mTOR pathway inhibitors and their application in personalized therapy for non-small-cell lung cancer. J Thorac Oncol. 2012 Aug;7(8):1315-26. doi: 10.1097/JTO.0b013e31825493eb. PMID: 22648207.
- Saliani M, Jalal R, Ahmadian MR. From basic researches to new achievements in therapeutic strategies of KRAS-driven cancers. Cancer Biol Med. 2019 Aug;16(3):435-461. doi: 10.20892/j.issn.2095-3941.2018.0530. PMID: 31565476; PMCID: PMC6743616.
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006 Jun 16;125(6):1137-49. doi: 10.1016/j.cell.2006.05.013. PMID: 16777603.
- Liu SV. NRG1 fusions: Biology to therapy. Lung Cancer. 2021 Aug;158:25-28. doi: 10.1016/j.lungcan.2021.05.011. Epub 2021 May 11. PMID: 34098222.
- Dong Hoon Shin, Lee D, Dong Ho Hong, Seung Hyun Hong, Hwang JA, Byung Il Lee, et al. Oncogenic function and clinical implications of SLC3A2-NRG1 fusion in invasive mucinous adenocarcinoma of the lung. Oncotarget. 2016 Sep 8;7(43):69450–65.
- Fernandez-Cuesta L, Plenker D, Osada H, Sun R, Menon R, Leenders F, Ortiz-Cuaran S, Peifer M, Bos M, Daßler J, Malchers F, Schöttle J, Vogel W, Dahmen I, Koker M, Ullrich RT, Wright GM, Russell PA, Wainer Z, Solomon B, Brambilla E, Nagy-Mignotte H, Moro-Sibilot D, Brambilla CG, Lantuejoul S, Altmüller J, Becker C, Nürnberg P, Heuckmann JM, Stoelben E, Petersen I, Clement JH, Sänger J, Muscarella LA, la Torre A, Fazio VM, Lahortiga I, Perera T, Ogata S, Parade M, Brehmer D, Vingron M, Heukamp LC, Buettner R, Zander T, Wolf J, Perner S, Ansén S, Haas SA, Yatabe Y, Thomas RK. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov. 2014 Apr;4(4):415-22. doi: 10.1158/2159-8290.CD-13-0633. Epub 2014 Jan 27. PMID: 24469108.
- Thiele CJ, Li Z, McKee AE. On Trk--the TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin Cancer Res. 2009 Oct 1;15(19):5962-7. doi: 10.1158/1078-0432.CCR-08-0651. Epub 2009 Sep 15. PMID: 19755385; PMCID: PMC2756331.
- Ricci A, Greco S, Mariotta S, Felici L, Bronzetti E, Cavazzana A, Cardillo G, Amenta F, Bisetti A, Barbolini G. Neurotrophins and neurotrophin receptors in human lung cancer. Am J Respir Cell Mol Biol. 2001 Oct;25(4):439-46. doi: 10.1165/ajrcmb.25.4.4470. PMID: 11694449.
- Chen B, Liang Y, He Z, An Y, Zhao W, Wu J. Autocrine activity of BDNF induced by the STAT3 signaling pathway causes prolonged TrkB activation and promotes human non-small-cell lung cancer proliferation. Sci Rep. 2016 Jul 26;6:30404. doi: 10.1038/srep30404. PMID: 27456333; PMCID: PMC4960652.
- Uren RT, Turnley AM. Regulation of neurotrophin receptor (Trk) signaling: suppressor of cytokine signaling 2 (SOCS2) is a new player. Front Mol Neurosci. 2014 May 14;7:39. doi: 10.3389/fnmol.2014.00039. PMID: 24860421; PMCID: PMC4030161.